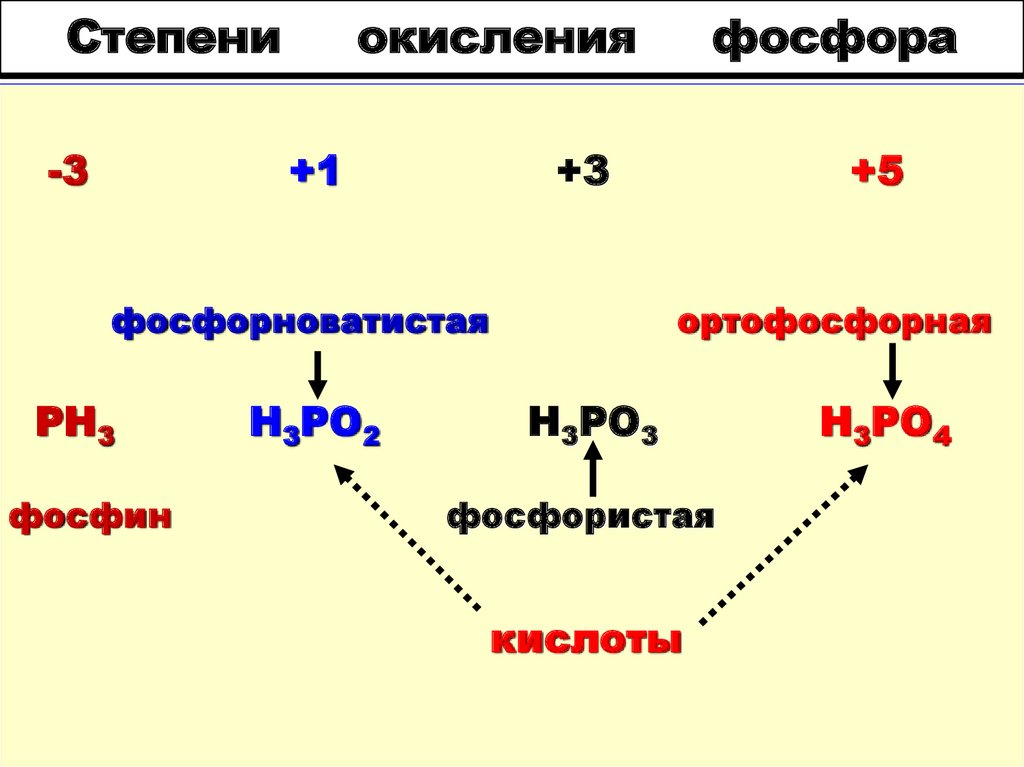
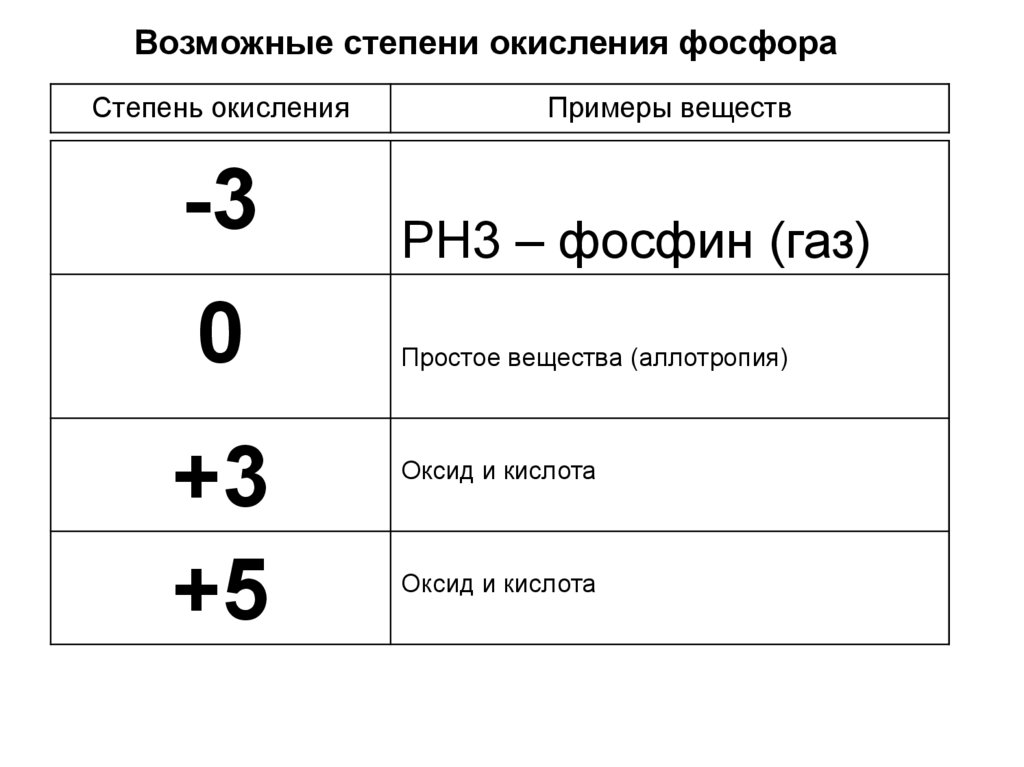
Фосфин: получение и химические свойства
Фосфин
Строение молекулы и физические свойства
Фосфин PH3 – это бинарное соединение водорода с фосфором, относится к летучим водородным соединениям. Следовательно, фосфин газ, с неприятным запахом, бесцветный, мало растворимый в воде, химически нестойкий и ядовитый. Водородные связи между молекулами фосфина не образуются. В твердом состоянии имеет молекулярную кристаллическую решетку.
Геометрическая форма молекулы фосфина похожа на структуру аммиака — правильная треугольная пирамида. Но валентный угол H-P-H меньше, чем угол H-N-H в аммиаке и составляет 93,5о.
У атома фосфора в фосфине на внешнем энергетическом уровне остается неподеленная электронная пара. Эта электронная пара оказывает значительное влияние на свойства фосфина, а также на его структуру. Электронная структура фосфина — тетраэдр , с атомом фосфора в центре.
Электронная структура фосфина — тетраэдр , с атомом фосфора в центре.
Способы получения фосфина
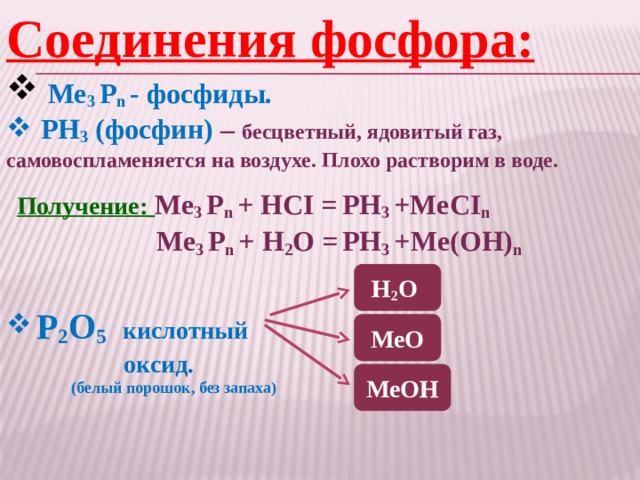
В лаборатории фосфин получают водным или кислотным гидролизом фосфидов – бинарных соединений фосфора и металлов.
Например, фосфин образуется при водном гидролизе фосфида кальция:
Ca3P2 + 6H2O → 3Са(ОН)2 + 2PH3
Или при кислотном гидролизе, например, фосфида магния в соляной кислоте:
Mg3P2 + 6HCl → 3MgCl2 + 2PH3↑
Еще один лабораторный способ получения фосфина – диспропорционирование фосфора в щелочах.
Например, фосфор реагирует с гидроксидом калия с образованием гипофосфита калия и фосфина:
4P + 3KOH + 3H2O → 3KH2PO2 + PH3↑
Химические свойства фосфина
1. В водном растворе фосфин проявляет очень слабые основные свойства (за счет неподеленной электронной пары). Принимая протон (ион H+), он превращается в ион фосфония. Основные свойства фосфина гораздо слабее основных свойств аммиака. Проявляются при взаимодействии с безводными кислотами.
В водном растворе фосфин проявляет очень слабые основные свойства (за счет неподеленной электронной пары). Принимая протон (ион H+), он превращается в ион фосфония. Основные свойства фосфина гораздо слабее основных свойств аммиака. Проявляются при взаимодействии с безводными кислотами.
Например, фосфин реагирует с йодоводородной кислотой:
PH3 + HI → PH4I
Соли фосфония неустойчивые, легко гидролизуются.
2. Фосфин PH3 – сильный восстановитель за счет фосфора в степени окисления -3. На воздухе самопроизвольно самовоспламеняется:
2PH3 + 4O2 → P2O5 + 3H2O
PH3 + 2O2 → H3PO4
3. Как сильный восстановитель, фосфин легко окисляется под действием окислителей.
Как сильный восстановитель, фосфин легко окисляется под действием окислителей.
Например, азотная кислота окисляет фосфин. При этом фосфор переходит в степень окисления +5 и образует фосфорную кислоту.
PH3 + 8HNO3 → H3PO4 + 8NO2 + 4H2O
Серная кислота также окисляет фосфин:
PH3 + 3H2SO4 → H3PO4 + 3SO2 + 3H2O
С фосфином также реагируют другие соединения фосфора, с более высокими степенями окисления фосфора.
Например, хлорид фосфора (III) окисляет фосфин:
2PH3 + 2PCl3 → 4P + 6HCl
Понравилось это:
Нравится Загрузка…
Осторожно — фосфин!
Э. С. Илларионова,
агрохимик, кандидат сельскохозяйственных наук (Дом ученых Пущинского научного центра РАН)
«Экология и жизнь» №6, 2008
Рассказ о газообразных соединениях фосфора, и прежде всего о фосфине, наверное, стоило бы начать со слов: «мерцающий свет, появляющийся на болотах (знаменитые «блуждающие огни») — результат самопроизвольного воспламенения фосфина». Ну а следующее определение — уже энциклопедического толка: «фосфин, или фосфористый водород (PH3) — это бесцветный газ с неприятным запахом (гниющей рыбы, чеснока или промышленного карбида), ядовит, образуется при биохимическом восстановлении эфиров фосфорной кислоты, преимущественно в анаэробных условиях, т. е. без доступа кислорода».
Ну а следующее определение — уже энциклопедического толка: «фосфин, или фосфористый водород (PH3) — это бесцветный газ с неприятным запахом (гниющей рыбы, чеснока или промышленного карбида), ядовит, образуется при биохимическом восстановлении эфиров фосфорной кислоты, преимущественно в анаэробных условиях, т. е. без доступа кислорода».
Соединения фосфора в природе
В природе встречается множество и других газообразных фосфорорганических соединений, в молекулах которых атом фосфора P соединен с атомом углерода C. Их насчитываются тысячи. Многие из них входят в состав экосистем, в том числе в живые клетки растений и микроорганизмов. Самая большая группа соединений со связями C–P открыта лет пятьдесят назад именно в живых объектах.
Есть в почвах и фосфонаты — производные фосфорорганических соединений с сохранившимися связями C–Р. Их, правда, немного, не более 1–2% от содержащегося в органике фосфора, поэтому на пашне их можно выявить далеко не всегда, зато в болотистых почвах и на лугах их содержание повышается до 3–4%.
В обычных (аэробных) условиях природные соединения органического и минерального фосфора — это фосфаты (ортофосфаты). Их существует великое множество. Для органических фосфатов характерна связь C–О–Р, иными словами, углерод и фосфор соединяются через атом кислорода.
Одна из удивительных загадок природы состоит в том, что органические фосфаты в живых системах (например, в водорослях и микроорганизмах) синтезируются и разлагаются не произвольно, а по правилу «золотого сечения», подчиняясь определенному закону, описываемому знаменитым рядом чисел Фибоначчи (1, 1, 2, 3, 5, 8…), в котором каждый следующий член равен сумме двух предыдущих. Гармония природы непостижимым образом проявляется здесь в накоплении и расходовании в экосистемах энергии и вещества (в частности, фосфора), описываемых соотношением, которое приближенно дается классическим коэффициентом «золотого сечения» 1,618 (5/3, 8/5, 13/8 и т. д.), т. е. 62% упомянутых соединений должно связываться и аккумулироваться и только 38% — разрушаться или улетучиваться. Эти закономерности сказываются в дальнейшем и на накоплении гумуса, и на круговороте фосфора и азота, и на газообразных потоках, определяемых выбросами и «стоками» углекислого газа СО2, и на «дыхании» почвы (выделении СО2 и усвоении кислорода О2). На самом деле в природе наблюдаются колебания числовых значений этого соотношения в пределах 1,3–1,7. Но, как не раз отмечалось в трудах автора и других ученых, гораздо страшнее оказывается то, что главной причиной отклонений и даже нарушения этой закономерности стала антропогенная деятельность.
Эти закономерности сказываются в дальнейшем и на накоплении гумуса, и на круговороте фосфора и азота, и на газообразных потоках, определяемых выбросами и «стоками» углекислого газа СО2, и на «дыхании» почвы (выделении СО2 и усвоении кислорода О2). На самом деле в природе наблюдаются колебания числовых значений этого соотношения в пределах 1,3–1,7. Но, как не раз отмечалось в трудах автора и других ученых, гораздо страшнее оказывается то, что главной причиной отклонений и даже нарушения этой закономерности стала антропогенная деятельность.
Некоторые специалисты уже обратили внимание на то, что нас могут подстерегать новые опасности, если это отношение стремится к единице, т. е. накопление и разложение идут с одинаковой интенсивностью, как это происходит, например, в цикле углерода, где за счет «вмешательства» мировой экономики океан и биосфера поглощают ныне лишь половину выбросов углерода (а надо бы 62%).
Но вернемся к фосфину и его производным, иными словами, к тем фосфорорганическим соединениям, в которых вместе с фосфором и углеродом встречаются разные элементы (азот, сера, кремний, молибден и т. д.) и их комплексы. В благоприятных для роста микроорганизмов условиях (в частности, в условиях болот и тундры при наблюдаемом потеплении) фосфорорганические соединения разлагаются с помощью фермента (катализатора) C–Р-лиазы. Ныне он обнаружен у 9 групп бактерий, которые питаются фосфором, добывая его при расщеплении фосфорорганических соединений. А вот грибы и дрожжи, на которые в экосистемах приходится 50–70% всей микрофлоры, не расщепляют эти соединения. Наоборот, простейшие, моллюски и грибы их синтезируют. Грибы могут расти даже при довольно высоких концентрациях фосфина, только мицелий у них желтеет.
д.) и их комплексы. В благоприятных для роста микроорганизмов условиях (в частности, в условиях болот и тундры при наблюдаемом потеплении) фосфорорганические соединения разлагаются с помощью фермента (катализатора) C–Р-лиазы. Ныне он обнаружен у 9 групп бактерий, которые питаются фосфором, добывая его при расщеплении фосфорорганических соединений. А вот грибы и дрожжи, на которые в экосистемах приходится 50–70% всей микрофлоры, не расщепляют эти соединения. Наоборот, простейшие, моллюски и грибы их синтезируют. Грибы могут расти даже при довольно высоких концентрациях фосфина, только мицелий у них желтеет.
Применение, свойства, опасности
Фосфин ядовит (опасная концентрация, способная привести к летальному исходу, — 0,05 мг/л), а в концентрации 2000 мл/м3 (2 л/м3, или 2·10–3) он вызывает мгновенную смерть. С ним приходится сталкиваться прежде всего в сельском хозяйстве при обеззараживании зернохранилищ и защите от клещей и других вредителей при транспортировке урожая, особенно зерновых культур. Раньше его активно применяли против крыс и мышей в амбарах. В Австралии к его помощи прибегают даже в борьбе с чрезмерно быстро размножающимися кроликами. Кроме того, ряд гербицидов и инсектицидов содержат фосфорорганические соединения на основе фосфина и его производных. И, наконец, в последнее время с ним все чаще приходится иметь дело в связи с широкомасштабным уничтожением химического оружия, предусматривающим обезвреживание отравляющих фосфорорганических соединений зарина и зомана — производных фосфина.
Раньше его активно применяли против крыс и мышей в амбарах. В Австралии к его помощи прибегают даже в борьбе с чрезмерно быстро размножающимися кроликами. Кроме того, ряд гербицидов и инсектицидов содержат фосфорорганические соединения на основе фосфина и его производных. И, наконец, в последнее время с ним все чаще приходится иметь дело в связи с широкомасштабным уничтожением химического оружия, предусматривающим обезвреживание отравляющих фосфорорганических соединений зарина и зомана — производных фосфина.
Чистый фосфин (без примесей) воспламеняется при температуре 150°С, сгорает с образованием токсичной фосфорной кислоты, но при наличии примесей дифосфина Р2Н4 или газообразного фосфора Р4 может и самопроизвольно воспламеняться на воздухе. Реакция фосфина с кислородом (как, впрочем, и окисление похожих на него метана — СН4 и силана — SiH4) относится к разветвленным цепным химическим реакциям, т. е. протекает все быстрее и может привести к взрыву. Окисление фосфина происходит при комнатной температуре, но при низкой температуре газ может быть устойчивым. Окисление фосфина можно ускорить, облучая его ультрафиолетом. Его самовоспламенение на воздухе возможно при концентрациях 1,7–1,9% (17–19 л/м3), или 26–27 г/м3. Так что в болотных экосистемах нередко приходится сталкиваться не только с упомянутыми «блуждающими огнями», но и с самовозгоранием (кстати, и распространенные торфяные пожары имеют такую же природу).
Окисление фосфина происходит при комнатной температуре, но при низкой температуре газ может быть устойчивым. Окисление фосфина можно ускорить, облучая его ультрафиолетом. Его самовоспламенение на воздухе возможно при концентрациях 1,7–1,9% (17–19 л/м3), или 26–27 г/м3. Так что в болотных экосистемах нередко приходится сталкиваться не только с упомянутыми «блуждающими огнями», но и с самовозгоранием (кстати, и распространенные торфяные пожары имеют такую же природу).
Для фумигации (избавления хранилищ зерна и сельскохозяйственной продукции от клещей и иных вредителей) обычно используют фосфиды, в частности, соединения фосфора с металлами. Реагируя с влагой воздуха, фосфиды выделяют фосфин. Содержащие фосфиды таблетки и ленты раскладывают в хранилищах из расчета 9 г/т зерна или другой подлежащей долгому хранению продукции, добавляют их даже в яблоки. Считается, что при проветривании фосфин улетучивается, хотя по имеющимся в научной литературе данным в фуражном зерне поглощается до 13% ядовитого газа. Разве одно это обстоятельство не должно заставить относиться к такой «дезинфекции» с предельной осторожностью?!
Разве одно это обстоятельство не должно заставить относиться к такой «дезинфекции» с предельной осторожностью?!
Ныне для фумигации зерна при транспортировке и хранении разрешены к применению два соединения — метилбромин и метилфосфин, причем первое на порядок менее токсично (и эффективно), чем второе. Применяя последнее, молчаливо предполагают, что ядовитый фосфин после поглощения содержимым хранилища чудесным образом извлекается и улетучивается, отравив лишь клещей и других вредителей. Похоже, раньше было не принято задумываться над тем, насколько эта картина соответствует действительности. Между тем еще почти полвека назад было установлено, что метилфосфин (смесь двух газов — метана СН4 и фосфина РН3) чрезвычайно токсичен, почти как сам фосфин.
Метан и фосфин в биосфере
Не секрет, что выделяемый из болот метан считается одним из основных парниковых газов и остается предметом активных обсуждений и исследований в связи с проблемами глобального изменения климата. Увы, в России его концентрация в атмосфере определяется только на одной метеостанции (Териберка на Кольском полуострове). А ведь ее не мешало бы измерять и над сибирскими болотами!
Увы, в России его концентрация в атмосфере определяется только на одной метеостанции (Териберка на Кольском полуострове). А ведь ее не мешало бы измерять и над сибирскими болотами!
Как известно, в земных глубинах законсервированы огромные запасы метана (7·1011–3·1013 т), причем 4·1011 т из них — в арктической зоне вечной мерзлоты.* На суше метан содержится в органических соединениях болот, осадках и детритах, а в Мировом океане — в газогидратах, залегающих под дном, в условиях пониженных температур. В Докладе ООН по изменению климата эксперты сообщают, что в Сибири выделение метана из болот и вечной мерзлоты в последние годы стремительно растет. Максимальная эмиссия метана из тундровых почв достигается при 8–10°С, а при 5°С преобладает его окисление на СО2 и воду. Образуется же он во всех почвенных горизонтах. В результате недавних исследований выяснилось, что, к примеру, наша южная кустарниковая тундра (окрестности Воркуты) служила стоком углерода лишь два года из последних пяти.
Это довольно опасная тенденция, особенно если принять во внимание, что на долю нашей страны приходится 2/3 всех болот на Земле. Наши площади заболоченных земель превышают площадь всех сельскохозяйственных угодий: по данным на 2003 года, 343 млн га болот (из них не поросших лесом — 130 млн га) и 221 млн га сельскохозяйственных угодий (из них 123 млн га пашни).
А вот как оценили выделение метана сотрудники МГУ в 2007 году по результатам измерений на болотах в Томской области. По их оценкам, среднее значение величины потока метана составляло около 10 мг/м2 за час. В летний период за сутки может выделяться 2,4 кг/га, за сезон (6 месяцев) 432 кг/га. А со 130 млн га болот — почти 60 млн т. На окисление такого количества метана потребуется вдвое больше кислорода — 120 млн т.
Главным же «побочным» эффектом выделения метана следует признать тот факт, что в тундровых и болотных экосистемах при низких температурах метан не только представляет собой изрядный резерв углерода, способный заметно изменить его содержание в атмосфере, но и тесно связан с фосфорорганическими соединениями, которые неизменно присутствуют в растениях, микрофлоре болот и осадков (в основном за счет упомянутой связи С–Р). И его выделение из тех мест, где он прежде синтезировался, из-за интенсификации с ростом температуры биохимических процессов брожения происходит не в последнюю очередь за счет распада соединений на основе фосфина. Иными словами, эмиссия газов СН4 и РН3 происходит параллельно. Между тем пока экологи и климатологи следят лишь за изменением содержания в атмосфере СО2 и СН4, а содержание РН3 никем не учитывается. А зря!
И его выделение из тех мест, где он прежде синтезировался, из-за интенсификации с ростом температуры биохимических процессов брожения происходит не в последнюю очередь за счет распада соединений на основе фосфина. Иными словами, эмиссия газов СН4 и РН3 происходит параллельно. Между тем пока экологи и климатологи следят лишь за изменением содержания в атмосфере СО2 и СН4, а содержание РН3 никем не учитывается. А зря!
Это упущение объясняется, в частности, тем, что лишь немногие специалисты знают о методах, позволяющих измерить содержание в атмосфере фосфора в газообразном состоянии. Ведь даже в научном мире до сих пор бытует мнение, что фосфор в природе существует преимущественно в форме фосфатов и после гидролиза связей Р–О–Р, Р–О–С и даже Р–С превращается в твердое вещество. Потоки фосфора в атмосферу в виде летучих соединений типа РН3 считаются ничтожными и ими пренебрегают. Определение содержания фосфора, поступившего в атмосферу с фосфином, лишь привычными методами, используемыми для выявления фосфора в твердых соединениях, заметно искажает реальную картину круговорота фосфора в экосистемах. При этом игнорируется появление в атмосфере ядовитого и самовозгорающегося фосфина.
При этом игнорируется появление в атмосфере ядовитого и самовозгорающегося фосфина.
Фосфиновая угроза: простые оценки
Между тем простейшую количественную оценку выделения фосфина в экосистемах можно получить, изучая затопленные водой территории, имитирующие заливные луга или рисовые чеки. Как было установлено в проведенной еще в 1926 году в Московской сельскохозяйственной академии им. К. А. Тимирязева серии из шести опытов, выполнявшихся в строго контролируемых условиях, в газовую форму (фосфин) переходит 9,7 мг фосфора из 1 кг почвы за час. Не слишком сложный расчет дает 2,13 кг/га за сутки. Но ведь это почти столько же, сколько выделяется метана из болот! Стало быть, за сезон получаем 383 кг/га, а со всей площади безлесных болот (130 млн га) — около 50 млн т РН3. На его окисление до фосфорной кислоты по формуле
РН3 + 2O2 → Н3РO4
потребуется, как нетрудно видеть, вдвое больше кислорода — почти 100 млн т (для метана эти значения составляли 60 и 120 млн т соответственно).
Косвенным подтверждением выделения фосфина из почв служат и исследования потоков фосфора на рисовых чеках — от посадки до уборки урожая потери фосфора в затопленных почвах в 3–8 раз превышают его содержание в зерне и соломе. Максимальный вынос Р2O5 достигает 100 кг/га. Из почв органических соединений фосфора выводится в 4 раза больше, чем запасается в растениях. Общие потери фосфора из верхнего (20 см) слоя почв, по разным оценкам, составляют 960–2940 кг/га. Есть данные, свидетельствующие о том, что при выращивании риса на затопленных чеках в течение 32 лет из почвы теряется больше половины гумуса, а с ним, конечно же, выносятся азот и фосфор.
Это может происходить и за счет выделения их газообразных форм — аммиака (NH3) и фосфина (РН3). Давно известно, что по химическим свойствам они представляют собой химические структурные аналоги. Повторюсь, определение фосфора и азота только в минеральной форме, игнорирование газовых составляющих не отражает истинных процессов в экосистемах, особенно в анаэробных условиях. В частности, прямое подтверждение того, что в экосистемах болот вместе с метаном выделяется и фосфор, получено в недавних исследованиях.**
В частности, прямое подтверждение того, что в экосистемах болот вместе с метаном выделяется и фосфор, получено в недавних исследованиях.**
Возвращаясь же к рассуждениям о возможной недооценке содержания фосфина в атмосфере, следует заметить, что вполне ощутимый вклад могут вносить не только болота Севера или тропиков, но и обширные рисовые плантации (прежде всего в Индии, Китае, Японии и странах Юго-Восточной Азии).
В научной литературе встречаются данные о том, что с осадками на землю выпадает до 3,5 кг/га фосфора. Иными словами, это примерно лишь 1% того фосфора, который, по имеющимся оценкам, выносится из болотных систем или затапливаемых почв фосфином в атмосферу (383 кг/га), остальные 99%, похоже, быстро окисляются, осаждаются или разлагаются (например, в результате гидролиза) в приземных слоях воздуха, литосфере и биосфере, обеспечивая перераспределение фосфора на поверхности земли.
Конечно же фосфин, как и метан, есть в атмосфере, но надо признать, что цикл фосфора изучен гораздо хуже, чем круговорот азота или углерода. Высокоактивные соединения фосфора в присутствии кислорода быстро превращаются в нейтральные комплексы, «безобидные» фосфаты. Кроме того, в экосистемах фосфора, как правило, немного, т. е. он присутствует в низких концентрациях. Поэтому, повторю, попытки учитывать фосфор только в форме фосфатов могут вести к заметному искажению его истинной роли в экосистемах. А к чему может привести недооценка этой роли, хорошо видно, например, по необдуманно осушенным ранее болотам, легко воспламеняющимся в засушливые годы за счет метана (СН4), силана (SiH4) и фосфина (РН3).
Высокоактивные соединения фосфора в присутствии кислорода быстро превращаются в нейтральные комплексы, «безобидные» фосфаты. Кроме того, в экосистемах фосфора, как правило, немного, т. е. он присутствует в низких концентрациях. Поэтому, повторю, попытки учитывать фосфор только в форме фосфатов могут вести к заметному искажению его истинной роли в экосистемах. А к чему может привести недооценка этой роли, хорошо видно, например, по необдуманно осушенным ранее болотам, легко воспламеняющимся в засушливые годы за счет метана (СН4), силана (SiH4) и фосфина (РН3).
По результатам измерений на упомянутой выше метеостанции Териберка было установлено, что в 1990 году в атмосферу с территории России было выброшено 48,8 млн т метана (напомним, наши оценки для всей площади безлесных болот составили около 60 млн т). За 1996–2003 гг. самая высокая концентрация была зафиксирована именно в 2003 году. Этот год был самым теплым для всей России, особенно же это относилось к лету и осени в зонах болот и тундры (Якутия, Западная Сибирь) — в среднем температура здесь оказалась выше многолетней почти на 6°С. В этих условиях одновременно наблюдалось и летнее снижение содержания верхового озона O3 над Севером России на 5–10%. А ведь летом и здесь ускоряются процессы фотосинтеза и образования кислорода. Поэтому очевидно, что для окисления возросшего количества метана и фосфина в условиях теплого 2003 года здесь интенсивно расходовался озон.
В этих условиях одновременно наблюдалось и летнее снижение содержания верхового озона O3 над Севером России на 5–10%. А ведь летом и здесь ускоряются процессы фотосинтеза и образования кислорода. Поэтому очевидно, что для окисления возросшего количества метана и фосфина в условиях теплого 2003 года здесь интенсивно расходовался озон.
От фосфина к кислороду: немного статистики и философии
Не секрет, что из-за богатейших биоресурсов Россию уже привыкли считать всемирным донором кислорода. По оценкам специалистов, над ее территорией ежегодно формируется 8130 млн т O2. Думается, мы не слишком погрешим против истины, предположив, что и процесс фотосинтеза, ответственный за формирование этой массы кислорода, подчиняется упомянутому «закону всемирной гармонии» — правилу «золотого сечения». Ведь на образование 1 т органики при фотосинтезе тратится 1,47 т углекислого газа, 0,6 т воды и 3,84 Гкал солнечной энергии и при этом выделяется 1,07 т кислорода. Соотношение между количеством поглощенного СO2 и выделенного O2 (1,47 : 1,07) не так уж отличается от «золотого».
По некоторым опубликованным оценкам, потребление кислорода в России (дыхание, сжигание топлива и другие промышленные нужды) составляет 2784 млн т. Тогда его «производство» Россией превышает ее расход на 5346 млн т. Но в других расчетах, где учтено потребление кислорода микрофлорой (прежде всего почвы) на «дыхание», российский избыток выработки кислорода над его потреблением оказывается уже на порядок ниже — 560 млн т. Между тем, как считают некоторые исследователи, «дыхание» почвы регулируется своим правилом «золотого сечения», определяющим соотношение выделяемого микрофлорой углекислого газа и потребляемого кислорода. На целине значение этой величины близко к 1,58, а на пашне колеблется в пределах 1,3–1,75 — иными словами, кислород в процессе «дыхания» почвы расходуется «экономно» (42–37%), а углекислого газа выделяется больше (58–63%). Если исходить из среднего значения «золотого сечения» 1,52 для соотношения СO2 : O2, то при эмиссии СO2 из почв России 10 409 млн т кислорода на «дыхание» российских почв потребляется еще 6848 млн т (оценки 2004 года по данным сотрудников Института фундаментальных проблем биологии РАН, в частности В. Н. Кудеярова).
Н. Кудеярова).
Своеобразная «золотая пропорция» соблюдается и между стоком СO2 и его эмиссией в масштабе России. Соотношение между стоком, составляющим 4450 млн т за год (в пересчете на углерод), и эмиссией (2800 млн т — в тех же единицах) оказывается равным 1,59, т е. удивительно близко к «золотому». Что ж, пока над Россией в целом нет избытка СO2, наши экосистемы поглощают больше, чем мы выбрасываем, наши леса нас спасают и покрывают «грехи» наши. Но в последние годы (прежде всего на Севере) все чаще отмечается, что экосистемы не справляются с «планом» по поглощению и отмеченное соотношение нарушается.
Впрочем, гораздо важнее, что, как следует из ряда оценок, на территории России общий расход кислорода за год на наши нужды (2784 млн т), дыхание почвы (6848 млн т) и окисление метана и фосфина (220 млн т) приближается к 10 млрд т, а это почти на 2 млрд т больше, чем его вырабатывают все наши леса. И этот печальный баланс представляется мне гораздо более серьезной проблемой, чем ожидаемая торговля квотами. Ради сохранения окружающей среды и биосферы планеты, ресурсов которой мы сегодня расходуем на 25% больше, чем они успевают восстанавливаться, нужно наконец осознать, что без ограничения потребления нам и нашим потомкам просто не выжить. И не в последнюю очередь это касается кислорода. В атмосфере его вроде бы немало (21%), но нельзя допускать, чтобы на Земле его потреблялось больше, чем вырабатывается.
Ради сохранения окружающей среды и биосферы планеты, ресурсов которой мы сегодня расходуем на 25% больше, чем они успевают восстанавливаться, нужно наконец осознать, что без ограничения потребления нам и нашим потомкам просто не выжить. И не в последнюю очередь это касается кислорода. В атмосфере его вроде бы немало (21%), но нельзя допускать, чтобы на Земле его потреблялось больше, чем вырабатывается.
Подводя итоги
Не секрет, что за последние 100 лет в результате бездумной деятельности человека и игнорирования им законов природы выбросы углекислого газа в атмосферу (и его содержание там), по разным оценкам, выросли на 25–35%. Одним из плохо просчитываемых последствий глобального потепления может стать резкая интенсификация биохимических процессов в природных зонах болот и вечной мерзлоты. При этом может резко возрасти выделение не только метана (это уже почти очевидно), но и мало изученных по влиянию на биосферу газов: аммиака, силана и фосфина, которым для окисления и нейтрализации потребуется немало кислорода. А ведь есть еще и не вполне проанализированные эффекты обратной связи (например, более интенсивное выделение метана ускорит дальнейший рост концентрации СO2 в атмосфере, что, в свою очередь, может привести к резкому замедлению фотосинтеза). Как следует из недавних исследований, в 90-х годах прошлого века заметно ослабла компенсирующая роль фотосинтеза в бореальных лесах. А ведь прежде было твердо установлено, что деревья на всех широтах достоверно способствовали фотосинтезу и ассимиляции СO2. Опасная тенденция! И примеры подобных «метаморфоз» лесов множатся год от года.
А ведь есть еще и не вполне проанализированные эффекты обратной связи (например, более интенсивное выделение метана ускорит дальнейший рост концентрации СO2 в атмосфере, что, в свою очередь, может привести к резкому замедлению фотосинтеза). Как следует из недавних исследований, в 90-х годах прошлого века заметно ослабла компенсирующая роль фотосинтеза в бореальных лесах. А ведь прежде было твердо установлено, что деревья на всех широтах достоверно способствовали фотосинтезу и ассимиляции СO2. Опасная тенденция! И примеры подобных «метаморфоз» лесов множатся год от года.
В настоящее время мы почти ничего не знаем о выделении и окислении не раз упоминавшегося в этой статье силана (SiH4). Между тем все болотные растения, злаки и микроорганизмы богаты органическим кремнием. В торфе верховых болот — 43% SiO2, переходных — 28%, низинных — 21%. Пока есть лишь отрывочные данные о том, что силан в соединении с фосфином образует недостаточно исследованные комплексы — силилфосфины. Процессы выделения силана, его окисления и соединения с другими элементами нуждаются в серьезном изучении.
Процессы выделения силана, его окисления и соединения с другими элементами нуждаются в серьезном изучении.
И в заключение — выглядящий фантастическим сюжет, который должен заставить задуматься всех, кто еще не утратил эту способность. В приземном слое атмосферы в связи со стремительным ростом содержания углекислого и некоторых других «мертвых» газов в обозримом будущем может возникнуть нехватка кислорода не только из-за замедления фотосинтеза, роста потребления на окисление, сжигание и дыхание, но и из-за «экрана» ядовитых газов, мешающего притоку O2 из более высоких слоев атмосферы.
Миллиарды лет основой всего живого на Земле был фотосинтез, исправно снабжавший планету кислородом. Увы, как справедливо отмечают некоторые исследователи, современная цивилизация впервые в истории, похоже, ухитрилась замедлить пополнение атмосферы кислородом, а природу довела до точки бифуркации. Выдержит ли она?
* См., например: Елдышев Ю.Н. Виновник глобального потепления — метан? // «Экология и жизнь», 2007, № 11, с. 45; Изменение климата: факты и факторы // «Экология и жизнь», 2008, № 3, с. 44.
45; Изменение климата: факты и факторы // «Экология и жизнь», 2008, № 3, с. 44.
** См., например, статью Кравченко И.К. в журнале «Микробиология», № 6, 2007.
Фосфорорганические соединения — wikidoc
Содержание
- 1 Обзор
- 2 Фосфаны и фосфины
- 2.1 Синтез
- 2.2 Реакции
- 2.3 Фосфаны
- 3 Оксиды фосфина
- 4 Фосфонаты
- 5 Фосфиты и сложные эфиры фосфорной кислоты
- 6 Фосфораны
- 7 Фосфор кратные связи
- 8 См. также
- 9 Внешние ссылки
- 10 Ссылки
Обзор
Фосфорорганические соединения представляют собой химические соединения, содержащие углерод-фосфорные связи. Фосфорорганическая химия — соответствующая наука, изучающая свойства и реакционную способность фосфорорганических соединений. Фосфор делит группу 15 в таблице Менделеева с азотом и соединениями фосфора, а соединения азота имеют много общего. [1] [2]
[1] [2]
Фосфор может принимать степени окисления -3, -1, 1, 3 и 5. В химической литературе очень часто соединения со степенью окисления +3 или -3 группируются как имеющие ( III) степени окисления независимо от знака. В официальной и более описательной номенклатуре соединения фосфора идентифицируются по их координационному числу δ и их валентности λ. В этой системе фосфин представляет собой δ
Фосфаны и фосфины
Исходным соединением фосфанов является PH 3 , называемое фосфином в США и Великобритании и фосфаном в других странах. [3] Замена одного или нескольких протонов органическим остатком дает Ph4-xR x , органофосфин или органофосфан, опять же в зависимости от страны. Атом фосфора в фосфанах/фосфинах имеет формальную степень окисления −3 (δ 3 λ 3 ) и являются фосфорсодержащими аналогами простых аминов.
Часто используемым органическим фосфином является трифенилфосфин.
Барьер для инверсии также намного выше, чем в аминах, для процесса, подобного инверсии азота, и поэтому фосфины с тремя разными заместителями могут проявлять оптическую изомерию.
Основность фосфинов меньше, чем у соответствующих аминов, например, сам ион фосфония имеет pKa -14 по сравнению с 9,21 для иона аммония; триметилфосфоний имеет p K a 8,65 по сравнению с 9,76 у триметиламина; и трифенилфосфоний (p K a 11.2) является менее основным, чем трифениламмоний (p K a 19).
Амины и фосфины имеют неподеленную пару электронов, но с разницей. В то время как неподеленная пара в амине делит свои электроны при каждой возможности делокализации, например, в пиридине, атом фосфора в аналогичной конфигурации этого не делает.
Реакционная способность фосфинов соответствует реакционной способности аминов в отношении нуклеофильности при образовании солей фосфония общей структуры PR 4 + X − . Это свойство используется в реакции Аппеля, превращающей спирты в алкилгалогениды.
Различие в реакционной способности с аминами заключается в легкости окисления фосфинов до оксидов фосфинов.
Синтез
Синтетические продукты для фосфинов:
- Нуклеофильное замещение галогенидов фосфора металлоорганическими реагентами, такими как реактивы Гриньяра.
- Нуклеофильное замещение фосфидов металлов, образующееся в результате реакции металлического калия с фосфином, как в амиде натрия с алкилгалогенидами.
- Нуклеофильное присоединение фосфина к алкенам в присутствии сильного основания (часто КОН в ДМСО), применяются правила Марковникова.
 [4] Фосфин можно получить на месте из красного фосфора и гидроксида калия. Первичные (RPH 2 ) и вторичные фосфины (RRPH) не требуют основания с электронодефицитными алкенами, такими как акрилонитрилы.
[4] Фосфин можно получить на месте из красного фосфора и гидроксида калия. Первичные (RPH 2 ) и вторичные фосфины (RRPH) не требуют основания с электронодефицитными алкенами, такими как акрилонитрилы.
- Радикальное присоединение фосфинов к алкенам с ДАК или органическими пероксидами с образованием антимарковниковских аддуктов.
- Нуклеофильное присоединение фосфина и фосфинов к алкинам в присутствии основания. Вторичные фосфины реагируют с электронодефицитными алкинами, такими как фенилцианоацетилен без основания.
- Органическое восстановление оксидов фосфина, например, хлорсиланом.
Реакции
Основными типами реакций фосфинов являются:
- в качестве нуклеофилов, например, от алкилгалогенидов до солей фосфония. Фоспины являются ключевыми нуклеофильными катализаторами димеризации енонов в реакции Рауха-Курье.
- в качестве восстановителей:
- Фосфины являются восстановителями в реакции Штаудингера, превращающей азиды в амины, и в реакции Мицунобу, превращающей спирты в сложные эфиры.
 В этих процессах фосфин окисляется до оксида фосфина. Также было обнаружено, что фосфины восстанавливают активированные карбонильные группы, например, восстановление α-кетоэфира до α-гидроксиэфира в схема 2 . [5] В предлагаемом механизме реакции первый протон заимствован из метильной группы в триметилфосфине (трифенилфосфин не вступает в реакцию).
В этих процессах фосфин окисляется до оксида фосфина. Также было обнаружено, что фосфины восстанавливают активированные карбонильные группы, например, восстановление α-кетоэфира до α-гидроксиэфира в схема 2 . [5] В предлагаемом механизме реакции первый протон заимствован из метильной группы в триметилфосфине (трифенилфосфин не вступает в реакцию).
- При модификации подходящими заместителями, как в некоторых диазафосфоленах ( схема 3 ), полярность связи P-H может быть фактически инвертирована (см.: умполунг), и полученный гидрид фосфина может восстанавливать карбонильную группу, как в примере бензофенона еще одним способом. [6]
- Мультидентатные фосфины, такие как BINAP, являются важными лигандами в металлоорганической химии.
Фосфаны
Первичные фосфаны недостаточно используются в химии из-за их общей недостаточной устойчивости к кислороду. В одном исследовании [7] сообщается о нескольких новых стабильных на воздухе ароматических первичных фосфанах, полученных органическим восстановлением соответствующего фосфоната:
Стабильность объясняется конъюгацией между ароматическим кольцом и неподеленной парой фосфора.
Оксиды фосфина
Оксиды фосфина (обозначение δ 3 λ 3 ) имеют общую структуру R 3 P=O с формальной степенью окисления −1. Фосфины образуют водородные связи, поэтому многие фосфины растворимы в воде. Связь P = O очень полярна с дипольным моментом 4,51 D для трифенилфосфина.
Природа двойной связи фосфора с кислородом является предметом споров. Пятивалентный фосфор, как и азот, несовместим с правилом октета. В более старой литературе связь представлена как дательная связь, как оксид амина. Преобладает мнение о полной двойной связи с обратной связью между заполненной парой электронов кислорода и пустой d-орбиталью фосфора (отсутствующей в азоте). Проблема в том, что связь P=O не реагирует как любая двойная связь C=C, поскольку реакции присоединения отсутствуют, а участие d-орбитали фосфора в связывании не поддерживается in silico. Альтернативные теории отдают предпочтение ионной связи P + -O — , что само по себе должно объяснить короткую длину связи.
Фосфины легко окисляются до оксидов фосфина, что подтверждается направленным синтезом фосфакраун, фосфорного аналога азакраун [8] , где невозможно выделить сам фосфин. [9]
Фосфонаты
Фосфонаты имеют общую структуру R−P(=O)(OR) 2 . В реакции Хорнера-Уодсворта-Эммонса и гомологизации Зейферта-Гилберта фосфонаты используются в качестве стабилизированных карбанионов в реакциях с карбонильными соединениями. Фосфонаты имеют множество технических применений, а бисфосфонаты относятся к классу лекарств.
Фосфиты и сложные эфиры фосфорной кислоты
Сложные эфиры фосфитов или фосфиты имеют общую структуру P(OR)  Фосфиты используются в реакциях Перкова и Арбусова. Эфиры фосфорной кислоты с общей структурой P(=O)(OR) 3 и степенью окисления +5 имеют большое технологическое значение в качестве антипиренов и пластификаторов. Без связи Р-С эти соединения технически не являются фосфорорганическими соединениями.
Фосфиты используются в реакциях Перкова и Арбусова. Эфиры фосфорной кислоты с общей структурой P(=O)(OR) 3 и степенью окисления +5 имеют большое технологическое значение в качестве антипиренов и пластификаторов. Без связи Р-С эти соединения технически не являются фосфорорганическими соединениями.
Фосфораны
Фосфораны имеют степень окисления −5 (δ 5 λ 5 ) с исходным соединением нестабильным фосфораном PH 5 или λ 5 -фосфаном (лямбда-5-фосфан). Илиды фосфора представляют собой ненасыщенные фосфораны, используемые в реакции Виттига.
Фосфор с кратными связями
Существует много соединений с углерод-фосфорными кратными связями (P=C), таких как фосфаалкенов (R 2
C::PR) и фосфаалкинов (RC:::PR). В соединении фосфорина один атом углерода у бензола замещен фосфором. Реакционную способность фосфаалкенов часто сравнивают с реакционной способностью алкенов, а не с иминами. Причина в том, что ВЗМО фосфаалкенов — это не неподеленная пара фосфора (как в иминах — неподеленная пара амина), а двойная связь. Поэтому, как и алкены, фосфаалкены участвуют в реакциях Виттига, реакциях Петерсона, перегруппировках Коупа и реакциях Дильса-Альдера.
Причина в том, что ВЗМО фосфаалкенов — это не неподеленная пара фосфора (как в иминах — неподеленная пара амина), а двойная связь. Поэтому, как и алкены, фосфаалкены участвуют в реакциях Виттига, реакциях Петерсона, перегруппировках Коупа и реакциях Дильса-Альдера.
Первый фосфаалкен был синтезирован в 1974 году Беккером в виде кето-енольной таутомерии, похожей на перегруппировку Брука:
, где R = метил или фенил, а tms означает триметилсилил.
В том же году Гарольд Крото спектроскопически установил, что термолиз Me 2 PH дает CH 2 = PMe.
Общий метод синтеза фосфаалкенов заключается в 1,2-отщеплении подходящих прекурсоров, инициированном термически или основанием, таким как DBU, DABCO или триэтиламин:
Метод Беккера используется для синтеза фосфорной подвески поли(п-фениленвинилена): [10]
Дифосфены представляют собой соединения, содержащие фосфорные двойные связи P=P. Фосфазены имеют двойную связь P=N.
См. также
- Фосфин — PR 3
- Оксид фосфина — OPR 3
- Фосфинит — P(OR)R 2
- Фосфонит — P(OR) 2 R
- Фосфит — P(OR) 3
- Фосфинат — OP(OR)R 2
- Фосфонат — OP(OR) 2 R
- Фосфат — OP(OR) 3
Template:ChemicalBondsToCarbon
Внешние ссылки
- Фосфорорганическая химия @ http://users.ox.ac.uk Ссылка
Ссылки
- ↑ Dillon, K.B.; Мэти, Ф .; Никсон, Дж. Ф. Фосфор. Копия под копирку ; Джон Вили и сыновья, 1997. ISBN 0-471-97360-2
- ↑ Quin, LD Руководство по фосфорорганической химии ; Джон Вили и сыновья, 2000. ISBN 0-471-31824-8
- ↑ Золотая книга: Ссылка
- ↑ Арбузова С. Н.; Гусарова, Н. К.; Трофимов Б. А. «Нуклеофильные и свободнорадикальные присоединения фосфинов и фосфинхалькогенидов к алкенам и алкинам».
 Аркивок 2006 , часть v, 12–36 (EL-1761AR). Статья
Аркивок 2006 , часть v, 12–36 (EL-1761AR). Статья
- ↑ Чжан, В.; Ши, М. «Восстановление активированных карбонильных групп алкилфосфинами: образование α-гидроксиэфиров и кетонов». Хим. коммун. 2006 , 1218–1220. дои: 10.1039/b516467b
- ↑ Берк, С.; Гудат, Д.; Нигер, М.; Дю Мон, В.-В. « P — Замещенные водородом 1,3,2-диазафосфолены: молекулярные гидриды». Дж. Ам. хим. соц. 2006 , 128 , 3946–3955. дои: 10.1021/ja057827j
- ↑ Укрощение функциональной группы: создание стабильных на воздухе хиральных первичных фосфанов 2006 doi:10.1002/anie.200602143
- ↑ Эдвардс, П. Г.; Хей Р.; Ли, Д.; Ньюман, П. Д. «Матричный синтез 1,4,7-трифосфациклононанов». Варенье. хим. соц. 2006 , 128 , 3818–3830. дои: 10.1021/ja0578956
- ↑ На этапе 1 дифосфиноэтан координируется с ферроценом, содержащим дополнительные лиганды монооксида углерода и ацетонитрильный лиганд.
 Следующим этапом является гидрофосфинирование тривинилфосфином с последующим алкилированием этилбромидом и гидрированием водородом над палладием на угле. На последнем этапе шаблон железа удаляется бромом, но окисление фосфиновых групп неизбежно.
Следующим этапом является гидрофосфинирование тривинилфосфином с последующим алкилированием этилбромидом и гидрированием водородом над палладием на угле. На последнем этапе шаблон железа удаляется бромом, но окисление фосфиновых групп неизбежно.
- ↑ Фосфор Копии PPV: сопряженные полимеры и молекулы, состоящие из чередующихся фениленовых и фосфалкеновых фрагментов Винсент А. Райт, Брайан О. Патрик, Селин Шнайдер и Дерек П. Гейтс J. Am. хим. соц.; 2006 ; 128(27), стр. 8836-8844; (Статья) doi:10.1021/ja060816l
Шаблон: Исходники WikiDoc
Периодическая таблица WebElements » Фосфор » фосфин
- Формула: PH 3
- Формула системы Hill: H 3 P 1
- Регистрационный номер CAS: [7803-51-2]
- Вес формулы: 33,998
- Класс: гидрид
- Цвет: бесцветный
- Внешний вид: газ
- Температура плавления: -133°С
- Температура кипения: -88°C
- Плотность: 1,5 кг·м -3 (газ)
Ниже приведены некоторые синонимы фосфина :
- фосфин
- гидрид фосфора(III)
- гидрид фосфора
- тригидрид фосфора
Степень окисления фосфора в фосфине равна 3 .
Синтез
Недоступно
Твердотельная структура
- Геометрия фосфора: 3 координата: пирамидальная
- Прототип конструкции:
Элементный анализ
В таблице показано процентное содержание элемента для PH 3 (фосфин).
| Элемент | % |
|---|---|
| Н | 8,89 |
| Р | 91.11 |
Изотопная схема для PH
3На приведенной ниже диаграмме показана расчетная изотопная картина для формулы PH 3 с наиболее интенсивным ионом, установленным на 100%.
Ссылки
Данные на страницах этих соединений собраны и адаптированы из основной литературы и нескольких других источников, включая следующие.
- Р.Т. Сандерсон в Chemical Periodicity , Reinhold, New York, USA, 1960.

- Н.Н. Гринвуд и А. Эрншоу в Chemistry of the Elements , 2-е издание, Butterworth, UK, 1997.
- Ф.А. Коттон, Г. Уилкинсон, К.А. Мурильо и М. Бохманн, в Advanced Inorganic Chemistry , John Wiley & Sons, 1999.
- А. Ф. Тротман-Дикенсон, (редактор) в Комплексная неорганическая химия , Пергамон, Оксфорд, Великобритания, 1973.
- Р.В.Г. Wyckoff, в Crystal Structures , том 1, Interscience, John Wiley & Sons, 1963.
- A.R.West в Базовая химия твердого тела Химия , John Wiley & Sons, 1999.
- А. Ф. Уэллс в Структурная неорганическая химия , 4-е издание, Оксфорд, Великобритания, 1975.
- Дж.Д.Х. Донней, (ред.) в Определяющие таблицы данных о кристаллах , монография ACA № 5, Американская кристаллографическая ассоциация, США, 1963.
- Д.Р. Лиде, (редактор) в справочнике по химии и физике Chemical Rubber Company , CRC Press, Бока-Ратон, Флорида, США, 77-е издание, 1996.