
4. Строение и функции РНК. Виды РНК
В клетке молекулы РНК содержат ядра, цитоплазма, а также органоиды, в которых происходит синтез белка: рибосомы, митохондрии, хлоропласты.
Рибонуклеиновая кислота (РНК) — биополимер, представляющий собой одну цепочку нуклеотидов. Мономеры (нуклеотиды) РНК состоят из пятиуглеродного сахара — рибозы, остатка фосфорной кислоты и азотистого основания.
Рис. \(1\). Строение нуклеотида РНК
Три азотистых основания в молекулах РНК такие же, как и у ДНК — аденин, гуанин, цитозин, а четвертым является урацил.
Рис. \(2\). Сравнение нуклеотидов ДНК и РНК
Образование полимера РНК происходит также, как и у ДНК — за счёт ковалентных связей между углеводом рибозой одного нуклеотида и фосфорной кислоты другого.
Рис. \(3\). Образование вторичной структуры РНК
Информационные РНК (иРНК) образуются в ядре на ДНК при участии фермента РНК-полимеразы.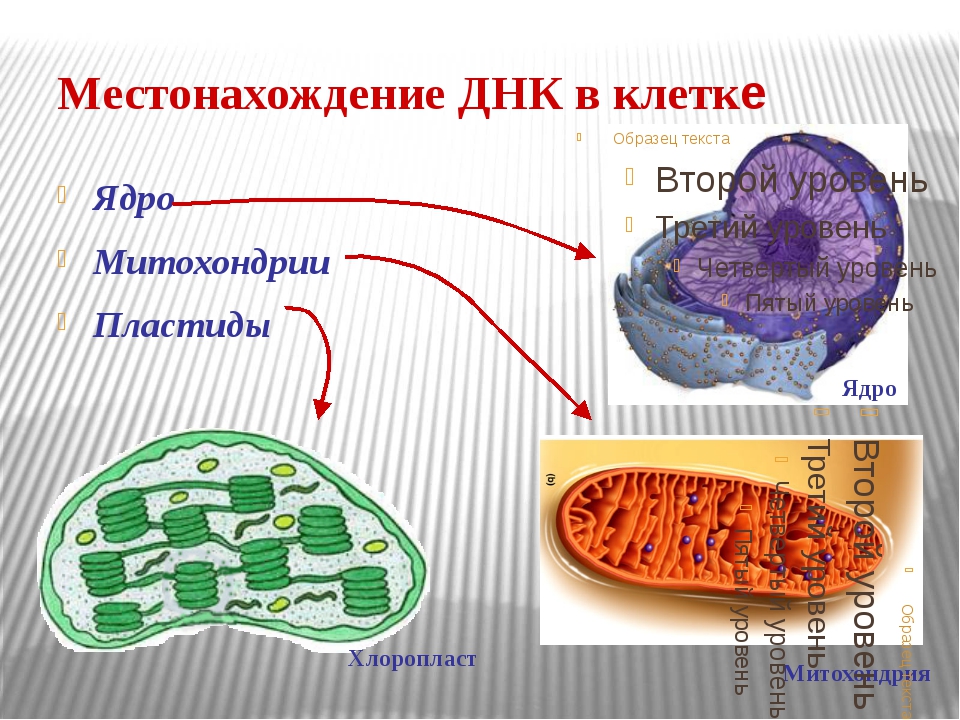 В клетке их содержание составляет приблизительно \(5\) % от всех РНК.
В клетке их содержание составляет приблизительно \(5\) % от всех РНК.
Функция иРНК — передача информации с ДНК к месту сборки белковых молекул, на рибосомы.
Рис. \(4\). Синтез РНК
Рибосомные РНК (рРНК) синтезируются в ядрышке и составляют основу рибосом, формируя активный центра рибосомы, в котором осуществляется биосинтез белка. рРНК составляют примерно \(85\) % всей РНК клетки.
Транспортные РНК (тРНК) тоже собираются в ядре, а затем перемещаются в цитоплазму. Они образованы небольшим количеством нуклеотидов (\(70\)–\(90\)). В клетке тРНК содержится примерно \(10\) %.
тРНК транспортируют аминокислоты к месту синтеза белка на рибосоме. Каждый вид аминокислот переносится отдельным видом тРНК.
Строение всех тРНК сходно. За счёт водородных связей некоторые участки молекул соединяются и образуются структуры, похожие на лист клевера. Отличаются молекулы тРНК тремя нуклеотидами, расположенными «на верхушке» структуры. Этот триплет (антикодон) комплементарен кодону иРНК, кодирующему соответствующую аминокислоту.
Отличаются молекулы тРНК тремя нуклеотидами, расположенными «на верхушке» структуры. Этот триплет (антикодон) комплементарен кодону иРНК, кодирующему соответствующую аминокислоту.
Аминокислота прикрепляется специальным ферментом к «черешку листа» и транспортируется в активный центр рибосомы.
Рис. \(5\). Транспортная РНК
Три вида РНК составляют общую функциональную систему, обеспечивающую реализацию генетической информации через синтез специфических для клетки белков.
Источники:
Рис. 1. Строение нуклеотида РНК © ЯКласс.
Рис. 2. Сравнение нуклеотидов ДНК и РНК © ЯКласс.
Рис. 3. Образование вторичной структуры РНК © ЯКласс.
Рис. 4. Синтез РНК © ЯКласс.
Рис. 5. Транспортная РНК © ЯКласс.
Росcийские биологи научились исследовать расположение ДНК в отдельной клетке
Ученые биологического факультета МГУ имени М.В. Ломоносова, Института биологии гена РАН и их коллеги из Австрии и США впервые построили подробные карты пространственной организации генома в индивидуальных клетках и изучили особенности пространственной организации материнского и отцовского геномов в зиготах мыши.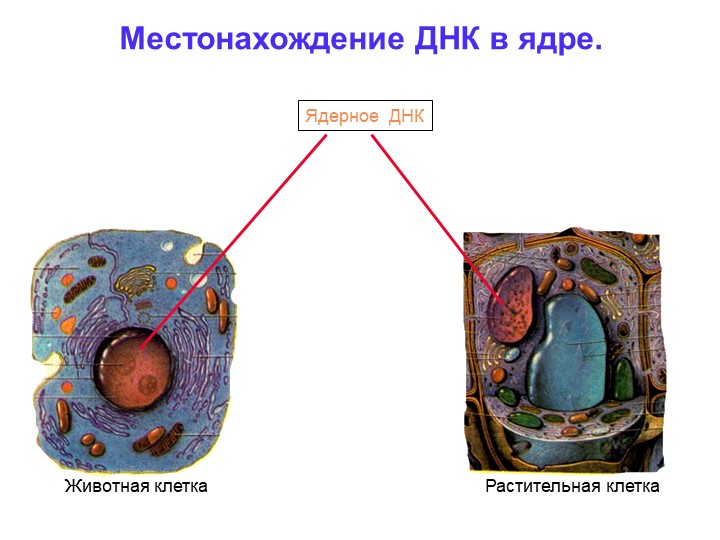 Результаты исследований опубликованы в журнале Nature.
Результаты исследований опубликованы в журнале Nature.
В ядрах клеток молекулы ДНК упакованы в особые структуры, хромосомы, которые можно представить себе как сложные, но не случайным образом спутанные клубки. Вещество хромосом, представляющее собой в основном комплекс ДНК, РНК и белков, называется хроматином. Биологи разработали новую методику изучения того, как хроматин упакован в ядре живой клетки. Эта методика является значительно усовершенствованным вариантом классического подхода к исследованию трехмерной структуры генома — Hi-C (high-throughput chromosome conformation capture).
«Возьмем три условных участка ДНК: А, B и С. Первые два расположены друг за другом в геноме — они соседи, а третий, предположим, находится от них на расстоянии в несколько миллионов пар нуклеотидов. Но хромосома может быть так упакована, что фрагмент С окажется рядом с А или В в пространстве. Мы можем установить этот факт (не для трех случайных участков ДНК, а в масштабе всего генома одновременно) и использовать эту информацию для построения карт пространственной структуры хроматина в живой клетке, так работает метод Hi-C», — рассказал один из авторов работы, кандидат биологических наук Сергей Ульянов.
В стандартном методе Hi-C для проведения одного эксперимента, как правило, требуется несколько сотен тысяч и даже миллионов клеток. Однако новая методика позволяет работать с одной отдельно взятой клеткой и составлять ее индивидуальную карту трехмерной структуры хромосом. Основным новшеством в этой методике является отбор единичных ядер на заключительном этапе Hi-C-эксперимента и проведение так называемой полногеномной амплификации — процесса, в котором с использованием особого фермента можно получить десятки тысяч копий ДНК из одного клеточного ядра.
«Мы провели анализ пространственной организации генома в зиготах мыши. Оказалось, что ядра, мужское и женское, которые сосуществуют в одной клетке, в зиготе, принципиально различаются по тому, как в них уложен геном. В ядре, сформировавшемся из ядра сперматозоида, активные участки генома в пространстве отделены от неактивных, а в ядре с материнским геномом этого не наблюдается. Во всех предыдущих исследованиях в клетках млекопитающих это разделение имело место, так что это очень неожиданный результат», — прокомментировал один из авторов статьи, Илья Флямер.
Пространственная организация хроматина является важным регуляторным инструментом, который клетка использует для управления экспрессией генов. В последнее время в научной литературе появляется все больше сообщений о том, что нарушения нормальной упаковки ДНК в ядре связаны с рядом тяжелых заболеваний человека и в первую очередь с некоторыми раковыми опухолями. Технология Hi-C на единичных клетках в будущем позволит исследовать отдельные, в том числе крайне немногочисленные, субпопуляции раковых клеток в составе опухолей и, возможно, приблизит нас к пониманию механизмов возникновения злокачественных новообразований.
Пресс-релизы о научных исследованиях, информацию о последних вышедших научных статьях и анонсы конференций, а также данные о выигранных грантах и премиях присылайте на адрес [email protected].
Новый метод покажет, как РНК регулируют активность генома — МФТИ
Международная группа ученых, в которую входит представитель ФИЦ биотехнологии РАН, ИОГЕН и МФТИ, разработала новый, более надежный метод для изучения контактов РНК с ДНК в ядре клеток. Метод поможет определить роль РНК в регуляции работы генов. Статья об исследовании опубликована в Nature Communication. Работа была поддержана Российским научным фондом (РНФ).
Метод поможет определить роль РНК в регуляции работы генов. Статья об исследовании опубликована в Nature Communication. Работа была поддержана Российским научным фондом (РНФ).
РНК и активность генов
Ранее считалось, что РНК — это лишь промежуточная стадия между ДНК и белком (рисунок 1а). Но когда научный мир начал описывать работу генома, оказалось, что далеко не все участки ДНК кодируют РНК. Более того, даже те, с которых считывается РНК, не обязательно кодируют белки. Функция большинства некодирующих РНК до сих пор неочевидна. В разных типах клеток должны работать разные гены и синтезироваться разные белки: в клетке мозга — одни, в клетке крови — другие. Это значит, что существуют факторы, которые влияют на активность генов. Теперь ученые начали понимать, что некодирующие РНК также являются одним из этих факторов.
Рисунок 1. а) Реализация генов: с ДНК считывается РНК, а с РНК — белок. б) В ядре клетки молекула ДНК упакована с помощью специальных белков в хроматин, из которого и состоит хромосома. Источники: Таблица генетического кода; Wiring Diagram Database, Diagram Of Chromatin
Источники: Таблица генетического кода; Wiring Diagram Database, Diagram Of Chromatin
Известно, что длинные некодирующие РНК взаимодействуют с хроматином — веществом, которое представляет собой молекулу ДНК, плотно упакованную с помощью белков (рисунок 1б). Хроматин может менять свою структуру: разворачиваться и сворачиваться, открывая гены для считывания или, наоборот, закрывая их. Если некодирующие РНК связываются с определенными участками хроматина, они могут влиять на его структуру и таким образом регулировать активность этих участков. Чтобы понимать, как регулируется активность генов и как это влияет на специализацию клетки, необходимо знать, какие некодирующие РНК с какими участками связываются.
Как это работает
Есть несколько методов, которые позволяют определять места, где взаимодействуют РНК и хроматин. Однако из-за ряда ограничений они пропускают много взаимодействий, к тому же некоторые из них требуют для анализа большого количества материала или разрушения клетки. Авторы работы разработали новый метод, который не разрушает клетку до закрепления контактов РНК и хроматина и показывает более высокую точность, — они назвали его RADICL-seq.
Авторы работы разработали новый метод, который не разрушает клетку до закрепления контактов РНК и хроматина и показывает более высокую точность, — они назвали его RADICL-seq.
Рисунок 2. а) Реакции, производимые в ядре клетки. Красным показана РНК, черным — ДНК, голубым — белки, синим — связующая молекула. Черная точка — молекула, позволяющая «выцепить» комплекс из раствора. Пояснения даны в тексте. b) Реакции, производимые в растворе: 1) удаляются белки, 2) достраивается вторая цепь, 3) обрезается до определяемого размера, 4) присоединяются последовательности для распознавания и 5) производится секвенирование. Источник:
В ядре клетки большинство РНК связаны с хроматином: РНК закрепляется в белках, которые связывают ДНК. Метод RADICL-seq заключается в следующем. В ядро добавляется фермент, который делает разрывы на ДНК и оставляет свободные концы, пригодные к сшивке. Также добавляется фермент, который разрушает свободные РНК и тем самым повышает точность определения контактов.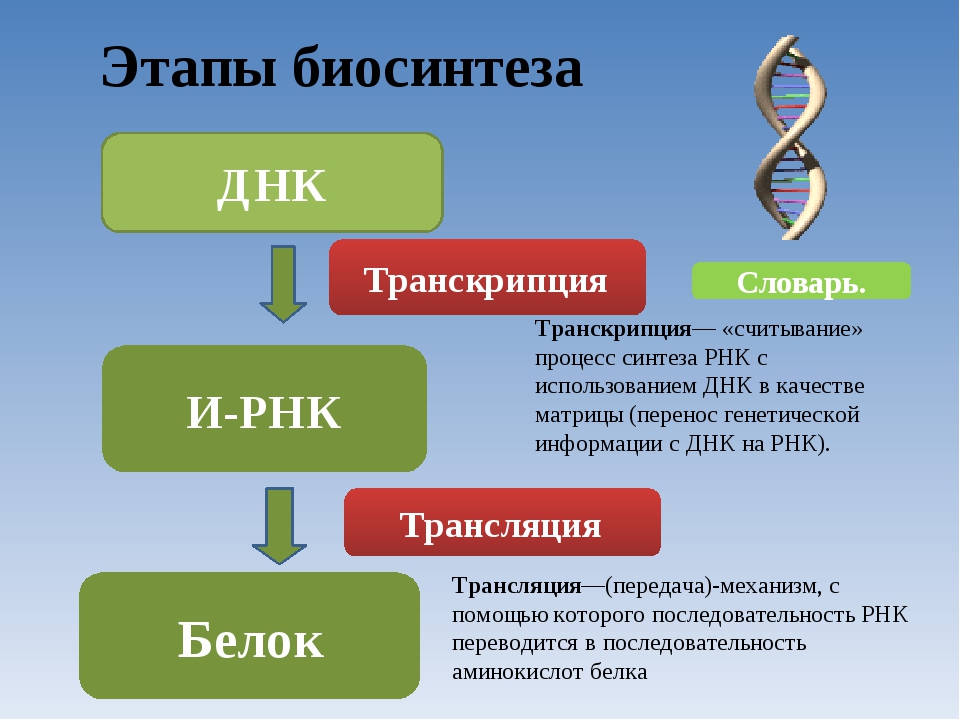 Потом добавляют молекулу, у которой один конец — одноцепочечный и связывается с РНК, а второй — двухцепочечный и связывается с расположенной рядом ДНК (рисунок 2а). Таким образом, эта молекула служит мостиком, скрепляющим РНК и ДНК. Дальше удаляют белки, достраивают вторую цепочку и получившийся ДНК-комплекс готовят к секвенированию (рисунок 2b), где определяют последовательности связанных РНК и ДНК.
Потом добавляют молекулу, у которой один конец — одноцепочечный и связывается с РНК, а второй — двухцепочечный и связывается с расположенной рядом ДНК (рисунок 2а). Таким образом, эта молекула служит мостиком, скрепляющим РНК и ДНК. Дальше удаляют белки, достраивают вторую цепочку и получившийся ДНК-комплекс готовят к секвенированию (рисунок 2b), где определяют последовательности связанных РНК и ДНК.
Раскодировать некодирующее
Ученые проверили метод RADICL-seq в действии. По сравнению с другими методами, он показал более высокую точность определения хроматин-РНК-взаимодействий. Благодаря высокому разрешению метода авторам удалось найти новые контакты не только некодирующих, но и кодирующих РНК с хроматином, включая те, которые расположены вдали от мест, где данная РНК считывается. Также они показали на клетках мыши, что метод подходит для изучения специфики взаимодействий в разных типах клеток. Они взяли две некодирующие РНК (одна из них, возможно, связана с шизофренией) и построили карту их взаимодействий с геномом в двух типах клеток: эмбриональных стволовых и предшественников олигодендроцитов (нейроглиальных клеток).
Исследование подтвердило важную роль длинных некодирующих РНК в регуляции участков генома, удаленных от мест считывания этих РНК. Гибкость метода RADICL-seq позволяет получить дополнительную биологическую информацию при внесении изменений в эксперимент. В частности, дает возможность обнаружить взаимодействия РНК-ДНК, не опосредованные белками хроматина. Наличие таких контактов указывает на роль в регуляции экспрессии генов не только канонических взаимодействий (таких как РНК-ДНК двойные спирали), но и неканонических (триплексов РНК-ДНК-), а также на значение некодирующих РНК в нацеливании белковых комплексов в конкретные места генома.
«Мы планируем дальше исследовать, как РНК участвует в регуляции экспрессии генов, архитектуры хроматина и, в конечном счете, на идентичность клеток. Вполне возможно, что в будущем с помощью этих некодирующих РНК можно будет контролировать активность конкретных генов, что важно, например, для лечения различных заболеваний»,
— прокомментировала Юлия Медведева, заведующая группой регуляторной транскриптомики и эпигеномики ФИЦ биотехнологии РАН и заведующая лабораторией биоинформатики клеточных технологий МФТИ, руководитель проекта по гранту РНФ.
Рисунок 3. Диаграммы, изображающие взаимодействия некодирующих РНК: Neat1 (a, b) и Fgfr2 (c, d) в эмбриональных стволовых клетках мыши (mESC) и клетках-предшественниках олигодендроцитов (mOPC). Neat1 синтезируется с 19-й хромосомы, а Fgfr2 — c 7-й. Источник: Nature Communication
Из чего собрана наша ДНК — Российская газета
Из школьного курса биологии вы наверняка помните, что ДНК — это нечто в форме спирали. В спираль закручены две нити. Если по биологии было отлично, то вспомните и сложную расшифровку аббревиатуры ДНК — дезоксирибонуклеиновая кислота. И независимо от оценки, при виде этой аббревиатуры на ум сразу приходит слово «ген». Но если б знали вы, из какого сора растут осмысленные гены и наши знания — о них и ДНК…
Научная догма
В 1953 году Джеймс Уотсон и Фрэнсис Крик опубликовали в журнале Nature двухстраничную статью с заголовком «Молекулярная структура дезоксирибонуклеиновых кислот».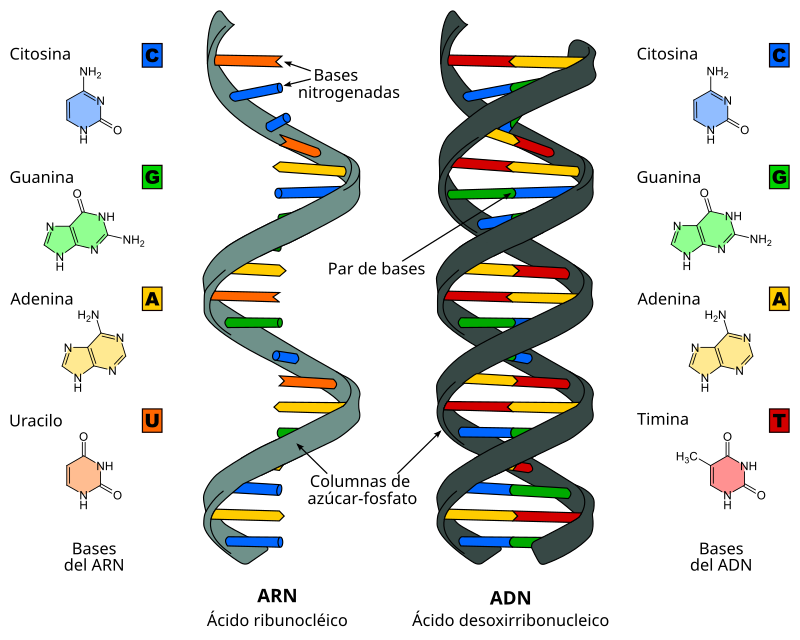 В статье коротенечко сообщалось, что ДНК — это двойная спираль, нити которой состоят из нуклеотидов, букв «генетического текста», и удерживаются вместе эфемерными водородными связями. Примерно тогда же стало понятно, как ДНК воспроизводит свои копии, и был сформулирован ключевой для всей биологии принцип — центральная догма* молекулярной биологии.
В статье коротенечко сообщалось, что ДНК — это двойная спираль, нити которой состоят из нуклеотидов, букв «генетического текста», и удерживаются вместе эфемерными водородными связями. Примерно тогда же стало понятно, как ДНК воспроизводит свои копии, и был сформулирован ключевой для всей биологии принцип — центральная догма* молекулярной биологии.
*Ее автор, один из первооткрывателей молекулярной структуры ДНК Фрэнсис Крик, объяснял выбор этого слова желанием подобрать звучное название.
Эта догма гласит: ДНК является средоточием генетической (наследственной) информации и может служить инструкцией — матрицей для синтеза своей ближайшей родственницы РНК, рибонуклеиновой кислоты. Последняя содержит информацию, которая используется для синтеза белков, а уже те самостоятельно принимаются за дело, выполняя огромное разнообразие работ в клетке. Вольный пересказ центральной догмы молекулярной биологии на сем окончен.
Появление этой догмы ознаменовало наступление «нового времени» в науке о живом. Но заметьте: что догма, что открытия 1950-х выхватили, словно лучом прожектора, только самый центральный, основополагающий сюжет с участием ДНК — кодирующие последовательности в генах. Все остальное первооткрыватели структуры чудо-молекулы пренебрежительно назвали «мусорной ДНК» (англ. Junk DNA), оставив без внимания.
Но заметьте: что догма, что открытия 1950-х выхватили, словно лучом прожектора, только самый центральный, основополагающий сюжет с участием ДНК — кодирующие последовательности в генах. Все остальное первооткрыватели структуры чудо-молекулы пренебрежительно назвали «мусорной ДНК» (англ. Junk DNA), оставив без внимания.
16 000 томов Толстого
В последующие десятилетия некодирующей «темной материи» не придавали особого значения. Но время шло, не стоял на месте и научный прогресс — ученые понемногу узнавали о разнообразии процессов, происходящих внутри ДНК и с ее участием. Немало удивительного удалось узнать и о геноме в целом. Например, что весь генетический код представляет собой длинный текст, который записан 4-буквенным алфавитом. Это так называемые нуклеотиды: аденин — A, тимин — T, гуанин — G и цитозин — C.
Не так давно, на рубеже тысячелетий, чтобы получить полную версию этого сакраментального «текста», был создан огромный международный консорциум «Геном человека». На протяжении более чем 10 лет исследователи из 20 научных центров США, Великобритании, Японии, Франции, Германии, Испании и Китая и нескольких частных компаний сплоченно работали и ежедневно докладывали о своих успехах.
На протяжении более чем 10 лет исследователи из 20 научных центров США, Великобритании, Японии, Франции, Германии, Испании и Китая и нескольких частных компаний сплоченно работали и ежедневно докладывали о своих успехах.
В результате огромной работы к 2003 году этот написанный природой и прочитанный человеком опус был наконец опубликован. В последовательности из 3 миллиардов букв* было найдено около 20-25 тысяч фрагментов — генов, — в которых непосредственно закодирована наследственная информация.
*Для сравнения: в 4-томном романе «Война и мир» всего-навсего около 750 тысяч знаков, включая знаки препинания и пробелы. Если разбить ДНК на отрезки, равные по числу знаков томам «Войны и мира», получится, что геном человека — это 16 000 таких томов.
Средняя длина гена — около 25-27 тысяч пар нуклеотидов. Если посчитать долю знаков всех генов от объема общего текста, получится около 2%. Если вычесть некодирующие элементы внутри генов, и того меньше. Но если в категорию «мусора» попало 98% генома, значит, этот мусор для чего-то да нужен?
Для сравнения: в 4-томном романе «Война и мир» всего-навсего около 750 тысяч знаков, включая знаки препинания и пробелы. Если разбить ДНК на отрезки, равные по числу знаков томам «Войны и мира», получится, что геном человека — это 16 000 таких томов.Проект «Геном человека» подарил много новых инструментов для работы с ДНК. Историки науки даже называют 2003 год началом новой эры в биологии — постгеномной. Менее чем за два десятилетия в арсенале учёных появились методы, позволяющие распознавать отдельный нуклеотиды при протягивании молекулы ДНК через нанопору, — в ХХ веке о таком не помышляли даже фантасты. И потихоньку наука начала разбираться с завалами «мусора».
Если разбить ДНК на отрезки, равные по числу знаков томам «Войны и мира», получится, что геном человека — это 16 000 таких томов.Проект «Геном человека» подарил много новых инструментов для работы с ДНК. Историки науки даже называют 2003 год началом новой эры в биологии — постгеномной. Менее чем за два десятилетия в арсенале учёных появились методы, позволяющие распознавать отдельный нуклеотиды при протягивании молекулы ДНК через нанопору, — в ХХ веке о таком не помышляли даже фантасты. И потихоньку наука начала разбираться с завалами «мусора».
Так из какого сора?
Что же представляет собой это «молчаливое большинство» нашего генома?
Безусловно, нельзя говорить о ненужности и бессмысленности 98% генетического материала. Эту хаотичную и слабо понятную сейчас массу можно назвать не мусором, а скорее свалкой сокровищ.
Некодирующие области могут выполнять разные функции или не выполнять никаких. Чтобы попасть в эту огромную категорию, участкам ДНК достаточно не хранить в себе информацию о структуре РНК или белка.
Компоненты человеческого генома
Кодирующие области 2%
Интроны 26%
ДНК-транспозоны 3%
LTR-ретротранспозоны 8%
LINEs 20%
SINEs 10%
Микросателлиты 3%
Другие типы 28%
Непосредственно внутри кодирующих областей встречаются интроны. Это такие участки ДНК, которые сидят внутри генов, но при этом ничего не кодируют. В дальнейшем интроны безжалостно вырезаются и выбрасываются из уже из РНК (этот процесс называется сплайсингом). Обилие подобных побочных продуктов производства РНК характерно для эукариот: у них для генома есть специальный контейнер, способный вместить сколь угодно много сора, — ядро. В человеческой ДНК на интроны приходится аж четверть текста. У бактерий такого контейнера нет, их геномы более компактные и рационализированные.
За границами кодирующих областей встречаются два типа крайне важных последовательностей: промоторы и терминаторы. Первые обозначают место, откуда надо начать считывание гена, вторые — конец.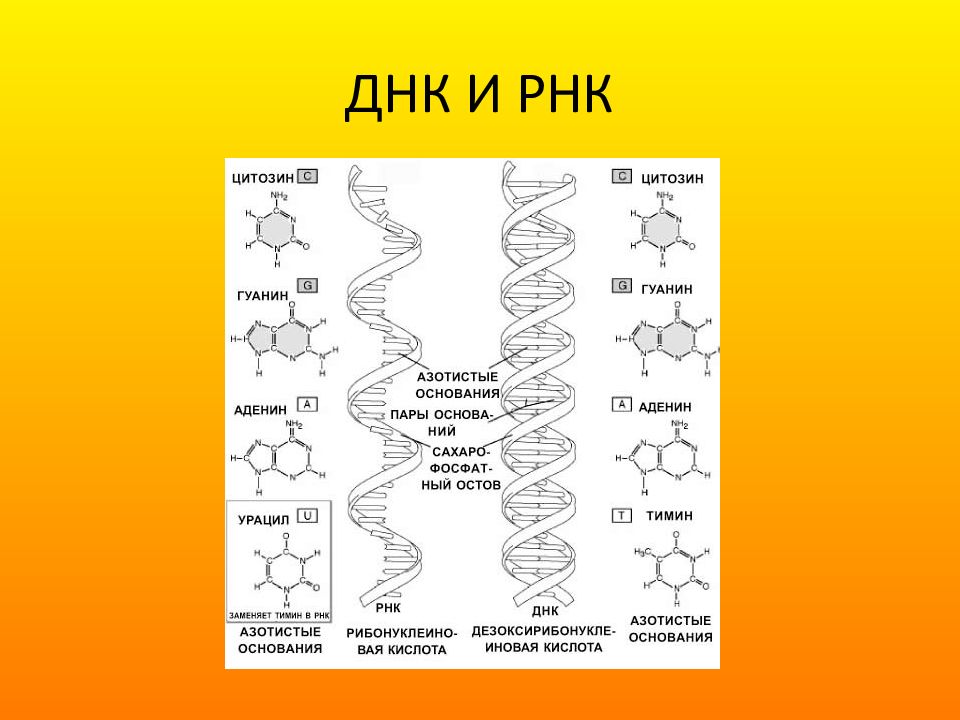 Рядом могут находиться энхансеры и сайленсеры — своеобразные тумблеры, позволяющие настроить активность считывания гена. Регуляторные участки ДНК — важный тип некодирующих последовательностей, ведь такая сложная машина, как организм, должна, во-первых, правильно собирать себя в процессе развития и, во-вторых, оперативно реагировать на изменения состояния — своего собственного и окружающей среды.
Рядом могут находиться энхансеры и сайленсеры — своеобразные тумблеры, позволяющие настроить активность считывания гена. Регуляторные участки ДНК — важный тип некодирующих последовательностей, ведь такая сложная машина, как организм, должна, во-первых, правильно собирать себя в процессе развития и, во-вторых, оперативно реагировать на изменения состояния — своего собственного и окружающей среды.
Плодятся буквы, как лопухи и лебеда, как буквы в ворде
«Ааааааааааааааааааааааааааааааааааааааааааааааааааа», — повторяет Владимир Сорокин несколько страниц в своем дебютном романе «Норма», используя бессмысленный повтор как средство художественной выразительности. TTTTTTTTTTTTTTTTTTTTT или GCAGCAGCAGCAGCAGCAGCAGCA, — вторит ему ДНК. На такие последовательности приходится около 1,5 миллиарда знаков из 3-миллиардного текста нашего генома. Почему бессмысленные повторы занимают столько места? Или, раз это место им отведено, они что-то да значат? Учёные считают, что повторяющиеся последовательности — это горячие точки эволюции: с ними связаны быстрые и неожиданные изменения генома. Исследования показали, что не все повторы одинаковы, их можно разделить на два больших типа: прямые и диспергированные, причём в каждом по несколько разновидностей.
Исследования показали, что не все повторы одинаковы, их можно разделить на два больших типа: прямые и диспергированные, причём в каждом по несколько разновидностей.
Диспергированные повторы, в отличие от прямых, не идут впритык друг за другом, а перемежаются с другими участками ДНК. По большей части диспергированные повторы — это транспозоны, последовательности-анархисты, способные скакать с места на место, то есть перемещаться по геному. Свободолюбивое поведение делает транспозоны важной движущей силой эволюции: они перемешивают и прочее население генома, вмешиваясь в работу генов.
Некоторые транспозоны — ретротранспозоны — могут не только скакать по геному, но и плодиться в нём. Их название отражает не художественные пристрастия, а механизм, который эти мобильные генетические элементы используют для передвижения по ДНК. Корень «ретро-» по аналогии с ретровирусами намекает на переход в форму РНК. То есть такие последовательности сначала считываются, а потом из РНК-матрицы переходят снова в ДНК, встраиваясь в новое место генома. Принцип «копировать — вставить». Оставшиеся малочисленные ДНК-транспозоны переходить в РНК не умеют, им остается вариант «вырезать — вставить».
Принцип «копировать — вставить». Оставшиеся малочисленные ДНК-транспозоны переходить в РНК не умеют, им остается вариант «вырезать — вставить».
Ретротранспозоны нашего генома можно разделить на несколько групп. Во-первых, это LTR-ретротранспозоны. На концах таких последовательностей присутствуют особые повторяющиеся участки. Подобные концевые повторы есть у ретровирусов и используются ими для встраивания генетического материала в геном хозяина. Это сходство названий и последовательностей не случайно: LTR-ретротранспозоны происходят от ретровирусов. Очередной вирусный след в человеческом геноме, притом увесистый: на LTR-повторы приходится порядка 8% генома Homo sapiens.
Ретротранспозоны LINEs и SINEs таких концевых повторов лишены. Главное различие между ними в размере: от менее 500 нуклеотидов у SINEs до в среднем 7000 у LINEs. Самые любопытные из них Alu-повторы. Подавляющее большинство SINEs генома Homo sapiens — это именно они. Предполагают, что Alu-элементы возникли около 100 млн лет назад и с тех пор изменялись вместе с расходящимися по собственной эволюционной ветке обезьянами. Изучение Alu-повторов помогает лучше понять родословную наших родичей.
Изучение Alu-повторов помогает лучше понять родословную наших родичей.
Ну сколько можно повторять!
Тандемные повторы ДНК вплотную примыкают друг к другу, почти как ездоки велосипеда-тандема. Отсюда их правильная структура с регулярным чередованием. Ну а свойства определяются размером повторов, по этому признаку тандемные повторы делят на три типа: сателлиты, минисателлиты и микросателлиты.
Самые длинные — сателлиты, или сателлитная ДНК, — могут тянуться миллионы и миллионы нуклеотидов подряд. Космическое название (англ. Satellite — спутник) связано с тем, что при ультрацентрифугировании (метод разделения веществ в результате очень быстрого раскручивания на ультрацентрифуге) эта часть ДНК легко отделялась от прочего генома. Сателлиты не кодируют РНК и белки и вообще складированы в «технических» областях хромосом: центромерах и теломерах. Центромеры и сателлитные повторы важны при делении клетки. Когда дело доходит до разделения удвоенных хромосом, микротрубочки подходят именно к центромерам и тянут за них хромосомы в противоположном направлении. А сателлиты в теломерах не позволяют концам хромосом слипаться (и самой хромосоме — разрушиться). Еще они защищают нас от старения, препятствуя укорачиванию хромосом.
А сателлиты в теломерах не позволяют концам хромосом слипаться (и самой хромосоме — разрушиться). Еще они защищают нас от старения, препятствуя укорачиванию хромосом.
Фото: Журнал «Кот Шрёдингера»
Средненькие в семье тандемных — минисателлиты, — как заведено и у людей, следуют за старшенькими. Они обнаружены не в самих центромерах и теломерах, а по соседству. Именно по последовательностям минисателлитов в детективных сериалах определяют сходство ДНК с места преступления с ДНК подозреваемого, а в романтических — устанавливают родительство. Длина каждого отдельного минисателлита довольно специфична у каждого из нас, однако у близких родственников они схожи.
Самые маленькие из тандемных повторов длиной всего 6-10 нуклеотидов — микросателлиты. Как и средненькие (и по тому же принципу), самые короткие в семействе нашли применение в криминалистике, но на этом их сходство со старшими братьями заканчивается. В отличие от них, микросателлиты находятся не в определённых участках хромосом, а распределены по всей ДНК почти равномерно.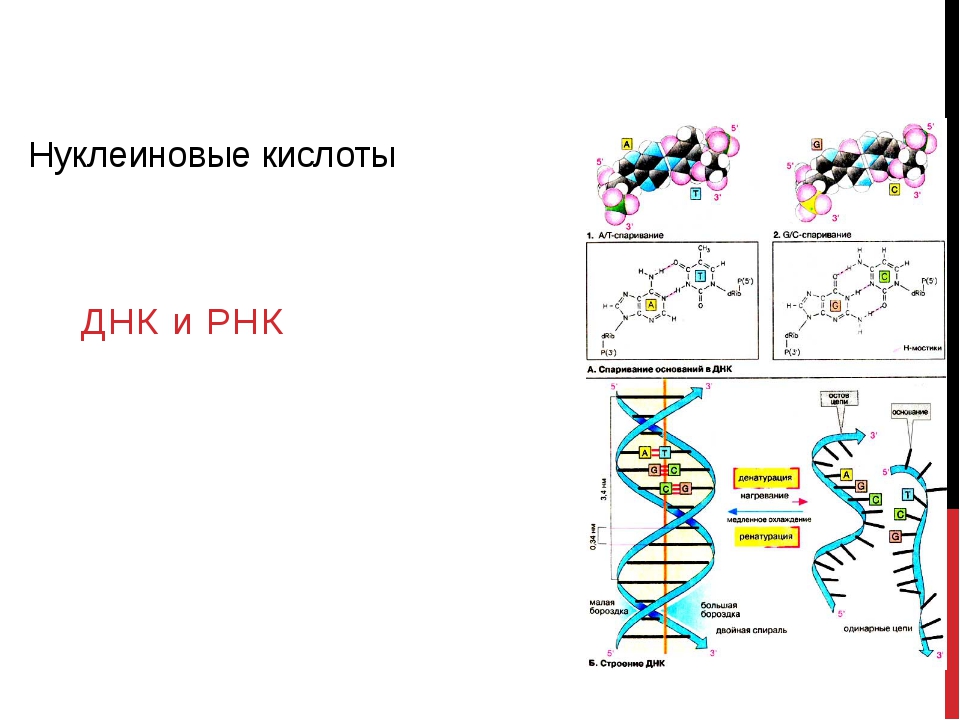 И не криминалистическое применение в них самое интересное. Микросателлиты, несмотря на малый размер, — кипучие котлы эволюции. Мутации в них происходят в тысячу раз чаще, чем в каких-либо других нуклеотидах. Характер этих мутаций заключается в потере или дублировании единиц-повторов целиком. Притом не одной — иногда «проскальзывание» копирующего ДНК белка затрагивает большие участки ДНК со следующими один за другим микросателлитами.
И не криминалистическое применение в них самое интересное. Микросателлиты, несмотря на малый размер, — кипучие котлы эволюции. Мутации в них происходят в тысячу раз чаще, чем в каких-либо других нуклеотидах. Характер этих мутаций заключается в потере или дублировании единиц-повторов целиком. Притом не одной — иногда «проскальзывание» копирующего ДНК белка затрагивает большие участки ДНК со следующими один за другим микросателлитами.
Если такое нарастание происходит в некоторых пределах, то в целом ситуация остается под контролем. Если же они превышены (к этому располагает наследственность), никто и ничто уже не сможет удержать микросателлиты. Происходит так называемая экспансия: единицы микросателлитов повторяются сотни и тысячи раз подряд, а обладатель соответствующего генома приобретает серьезное, обычно неизлечимое и быстро прогрессирующее заболевание. Это может быть и болезнь Хантингтона — неуклонно прогрессирующее заболевание мозга, прославившееся вместе с Тринадцатой из сериала «Доктор Хаус», и синдром хрупкой Х-хромосомы. Хрупкой оказывается как раз область микросателлитов, причем хрупкой настолько, что часть этой хромосомы может просто… отвалиться.
Хрупкой оказывается как раз область микросателлитов, причем хрупкой настолько, что часть этой хромосомы может просто… отвалиться.
Геном Homo sapiens — это текст из 3 миллиардов букв, который очень далек от порядка и предсказуемости. Наш геном — это разнообразие, хаос, повторы и… своеобразная эволюционная разумность. И неожиданные сюжетные повороты. Почти как в стихотворении Хармса об устройстве человека:
А, впрочем, не рук пятнадцать штук,
пятнадцать штук,
пятнадцать штук.
Хэу-ля-ля,
дрюм-дрюм-ту-ту!
Пятнадцать штук, да не рук.
Как работает ДНК?
Любой организм на Земле содержит в себе молекулярную инструкцию для собственной сборки и эксплуатации — это дезоксирибонуклеиновая кислота или ДНК. В ДНК закодированы самые разные черты: какой цвет глаз и волос будет у человека, какой у розы будет аромат или каким способом бактерия будет заражать хозяина. Атлас рассказывает, как код ДНК становится свойствами организма и передается по наследству, как «включаются» и «выключаются» гены и как упаковать длинную молекулу ДНК в крошечную клетку.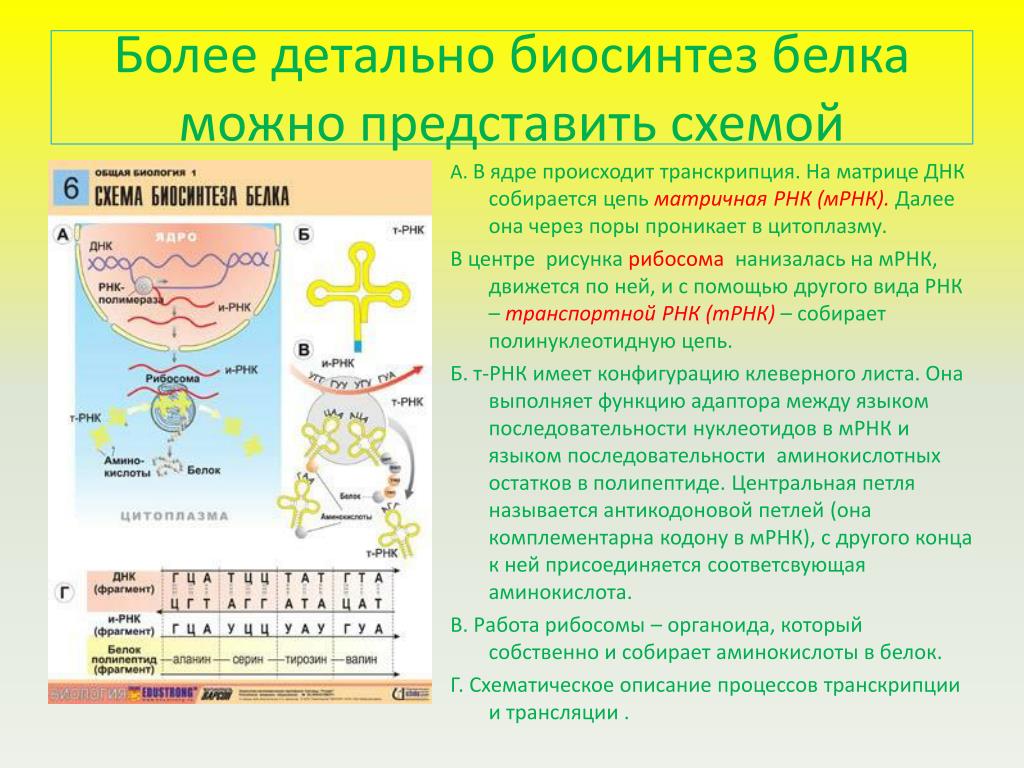
Содержание
Как устроена ДНК?
ДНК — это длинная и сложная молекула. Представить ее можно как двойную цепь, звенья которой — это нуклеотиды, состоящие из молекулы углевода (дезоксирибозы), остатка фосфора и азотистого основания. Нуклеотиды отличаются друг от друга по входящему в них азотистому основанию: Аденин, Тимин, Гуанин и Цитозин. Часто их обозначают просто буквами — А, Т, Г, Ц. Выстраиваясь друг за другом нуклеотиды образуют одну цепь ДНК. Порядок, в котором расположены нуклеотиды,отличается у разных организмов. ДНК-узор из А, Т, Г, Ц считывают другие молекулы и запускают формирование самых разных свойств организма.
В двойной цепи ДНК одна из цепей расположена как бы вверх ногами по отношению к другой — это расположение называется «антипараллельным».
Цепи в паре не идентичны друг другу, а сопоставимы по определенному правилу: если в первой цепи в последовательности А, во второй на этом же месте стоит Т, если в первой Г, то во второй-Ц, и наоборот.
Это правило называется принцип комплементарности. Запомните этот принцип, он чертовски важен для генов.
Между комплементарными нуклеотидами двух цепей возникают водородные связи, выстраивается как бы «лестничная структура» между цепями. Эта лестница ДНК закручена в двойную спираль.
Но в форме двойной спирали ДНК занимает слишком много места, у человека в длину она займет примерно 2 метра. Как уместить такую громадину в крохотное ядро? ДНК для этого скручивается и наматывается самыми разными способами. Для начала происходит наматывание ДНК на белки — гистоны, словно на катушки. Затем ДНК с гистонами переживает ещё один цикл «закручивания» в структуру, подобную плотной тугой спирали — соленоиду. Эта плотная нить в свою очередь укладывается петлями с помощью белков скаффолда, а затем складывается еще разок: в петлевые домены на белках матрикса ядра.
Когда спираль ДНК сматывается с помощью гистонов и белков скаффолда получается хроматида.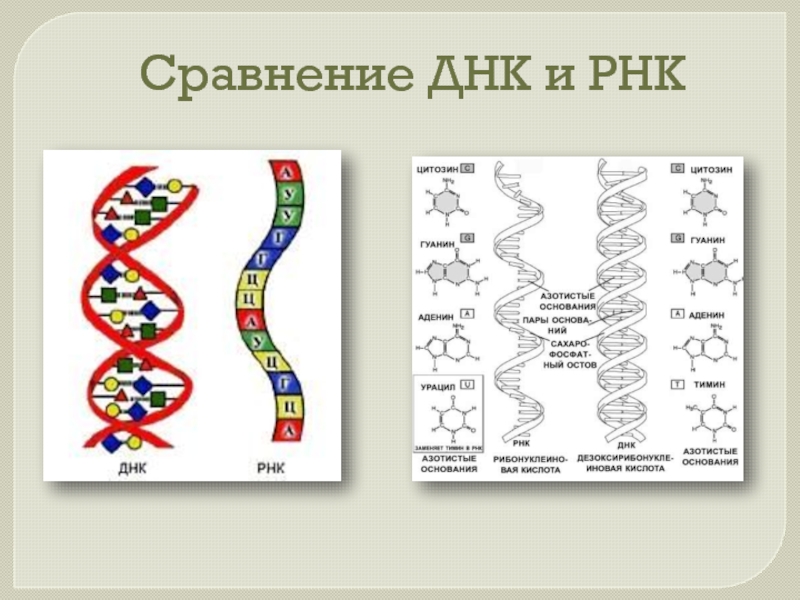 Две хроматиды, несущие одинаковую последовательностью ДНК, называются сестринские хроматиды. Одна или две хроматиды (их количество зависит от того, когда клетка делилась) образуют хромосому. В клетках нашего тела содержится 23 пары хромосом (то есть всего 46 штук).
Две хроматиды, несущие одинаковую последовательностью ДНК, называются сестринские хроматиды. Одна или две хроматиды (их количество зависит от того, когда клетка делилась) образуют хромосому. В клетках нашего тела содержится 23 пары хромосом (то есть всего 46 штук).
Как ДНК удваивается?
Когда клетка делится, каждая новая клетка хочет заполучить себе копию инструкции для жизни, поэтому ДНК приходится копировать. Удваивается ДНК полуконсервативно — это значит, что одна из цепей остается от исходной молекулы, а вторая синтезируется заново. С последовательности исходной, «материнской», ДНК по правилу комплементарности (А-Т; Г-Ц) можно переписать вторую, дочернюю, цепь ДНК.
Допустим, последовательность материнской цепи: ААГЦТТАГ, значит «дочерняя» будет комплементарна — ТТЦГААТЦ
А
— ТА
—ТГ—Ц
Ц —Г
Т
— АТ
—АГ—Ц
Две новые дочерние цепи строятся сразу с двух материнских цепей. Как это происходит? Для начала ДНК избавляется от гистонов. Затем фермент хеликаза расплетает двойную спираль ДНК и разрывает водородные связи между цепями. Фермент праймаза строит у каждой цепочки небольшую последовательность нуклеотидов — праймер. Праймер — это затравка для построения новой цепи ДНК. К праймерам присоединяется фермент ДНК-полимераза, который присоединяет новые нуклеотиды к строящимся цепочкам.
Как это происходит? Для начала ДНК избавляется от гистонов. Затем фермент хеликаза расплетает двойную спираль ДНК и разрывает водородные связи между цепями. Фермент праймаза строит у каждой цепочки небольшую последовательность нуклеотидов — праймер. Праймер — это затравка для построения новой цепи ДНК. К праймерам присоединяется фермент ДНК-полимераза, который присоединяет новые нуклеотиды к строящимся цепочкам.
Когда новые цепи синтезированы, между вновь синтезированной дочерней и исходной, материнской, цепочкой ДНК возникают водородные связи, вновь образуется двойная спираль, которая снова компактно сматывается. Когда клетка поделится, получившиеся ДНК-сёстры разойдутся по ядрам образовавшихся клеток.
Как информация ДНК превращается в свойства организма?
ДНК — это код, поэтому для начала ее надо расшифровать и переписать на язык другой нуклеиновой кислоты — рибонуклеиновой кислоты или РНК. Этот процесс называется транскрипция. РНК очень похожа на ДНК, однако вместо нуклеотидов с дезоксирибозой, в цепочку объединяются нуклеотиды с другим сахарным остатком — рибозой ( получаются рибонуклеотиды).
РНК словно под диктовку переписывается с последовательности ДНК так же, как при удвоении, да-да, вы догадались, по правилу комплементарности. Но есть одно исключение: в РНК азотистое основание Тимин заменено на основание Урацил: а значит, если в кодирующей ДНК А — то в РНК считается У. Присоединяет нуклеотиды РНК-полимераза.
Когда «буквенный» состав ДНК переписан в РНК происходит ее созревание: лишние части (интроны) вырезаются, а нужные (экзоны) сшиваются вместе. Получившаяся РНК — это инструкция к сборке белка. Читают эту инструкцию органеллы под названием рибосомы, а сам процесс называется трансляция.
Рибосомы насаживаются на начало цепи РНК и движутся по ней в направлении к ее концу. В ходе движения рибосомы читают РНК по слогам, а вернее, по триплетам или кодонам,то есть по трем нуклеотидам-буквам: АГЦ, АЦА, ААГ и так далее. Каждый такой триплет обозначает какую-то аминокислоту, как показано в таблице. Аминокислоты — это строительные кирпичики белков.
Аминокислоты — это строительные кирпичики белков.
УУУ — фенилаланин УУЦ — фенилаланин УУГ | УЦУ УАЦ — серин УЦА УЦГ | УАУ — Тирозин УАЦ УАА — стоп-кодон УАГ— стоп-кодон | УГУ — цистеин УГЦ УГА — стоп-кодон УГГ — триптофан |
ЦУУ ЦУЦ ЦУА — Лейцин | ЦЦУ ЦЦЦ ЦЦА — пролин ЦЦГ | ЦАУ —гистидин ЦАЦ ЦАА — глютамин ЦАГ | ЦГУ ЦГЦ — аргинин ЦГА ЦГГ |
АУУ | АЦУ АЦЦ АЦА —треонин АЦГ | ААУ— аспарагин ААГ | АГУ— серин АГЦ АГЦ — аргинин АГГ |
ГУУ | ГЦУ ГЦЦ ГЦГ | ГАУ —аспарагиновая кислота ГАЦ ГАА —глутаминовая кислота ГАГ | ГГУ ГГЦ — глицин ГГА ГГГ |

Когда кодон прочитан, рибосома понимает, какую аминокислоту надо присоединить, например, прочитала АЦА, — значит надо присоединить треонин, и вызывает соответствующий переносчик — транспортную РНК. Итак, рибосома движется по РНК, читает кодоны друг за другом и по о очереди вызывает тРНК с аминокислотами, которые навешиваются одна на другую. Получившаяся цепочка из аминокислот — это белок или его часть.
Получившаяся цепочка из аминокислот — это белок или его часть.
Почти все наши свойства определяют белки. Группу крови определяют белки на поверхности эритроцитов, белок фермент лактаза определяет вашу способность переваривать лактозу, а белок-рецептор дофамина может определять предрасположенность человека к рискованному поведению.Так послание ДНК, закодированное в А, Т, Г, Ц, через РНК, находит свое выражение в белке, который что-то делает и определяет свойства организма.
Почему во всех клетках есть полная последовательность ДНК, но часть генов не работают?
В каждой клетке нашего тела содержится полный набор генов. Но каким-то образом, разные клетки понимают, что одни должны стать волосами, другие костями, третьи мозгами. Для наших клеток важно не столько то, какие гены находятся в ДНК, сколько то, какие именно из них активны. Будет или не будет ген работать зависит от того, сможет ли РНК-полимераза его узнать и переписать с ДНК копию РНК. Если РНК-полимераза не связывается с геном, и не переписывает его — ген молчит.
Если РНК-полимераза не связывается с геном, и не переписывает его — ген молчит.
Чтобы привлечь РНК-полимеразу к гену существуют белки-активаторы: так называемые транскрипционные факторы. На гены транскрипционных факторов приходится очень много ДНК: всего в человеческом геноме закодировано около 20 000 белков, из них две тысячи — это транскрипционные факторы. То есть каждый десятый ген, по сути, сам ничего в клетке не производит, а лишь следит за тем, чтобы другие гены включились в нужное время в нужном месте.
Пример транскрипционного фактора — ген FOXP2. Если создать ГМО-мышей, которым внесли человеческую версию этого гена, то они лучше обучаются и пищат как-то иначе. Но ген вовсе не отвечает за строение голосовых связок или за особую активность нейронов. FOXP2 — это просто грамотный управляющий, транскрипционный фактор, который заставляет гены мыши работать как-то иначе.
Гены также можно заставить «замолкнуть». Например, есть белки-репрессоры, которые связываются с геном и не дают РНК-полимеразе присоединиться Другой способ выключить ДНК — модифицировать гистоны, то есть катушки, на которые намотана ДНК. Фермент деацетилаза гистонов заставляет ДНК наматывается на гистоны так туго, что никакие другие молекулы, ни полимеразы, ни факторы транскрипции, не могу к ней подобраться.
Фермент деацетилаза гистонов заставляет ДНК наматывается на гистоны так туго, что никакие другие молекулы, ни полимеразы, ни факторы транскрипции, не могу к ней подобраться.
«Выключает» гены и метилирование ДНК. Метильная группа присоединяется к цитозину с помощью фермента ДНК-метилтрансферазы. Такое метилирование происходит, если рядом с цитозином стоит гуанин — в составе так называемого динуклеотида CpG. Участки ДНК, богатые CpG-динуклеотидами, называют CpG-островками. Гиперметилирование CpG-островков приводит к конденсации — то есть сматыванию ДНК в плотные клубки: гены, попавшие в клубок, не читаются.
Такие внешние влияния на работу генов называют эпигенетикой. Эпигенетические факторы не меняют саму последовательность ДНК, но меняют активность генов. В последнее время показано, что эпигенетическими факторами, способными «включать»и «выключать» работу генов, являются работа в ночные смены, употребление алкоголя, курение, диета и физическая активность, и даже то, испытывали ли ваши родители стресс.

Согласно одному из исследований, холокост повлиял на геномы минимум двух поколений евреев произошло метилирование FKBP5 — гена, связанного с реакцией на стресс. В другом исследовании было показано, что у нидерландских детей, которые в утробе мамы пережили массовый голод в конце Второй мировой войны («Голодная зима») , даже спустя 60 лет оказался понижен уровень метилирования участка гена инсулин-подобного фактора роста IGF2 (участвующего в процессах роста и развития). Дети этих детей тоже были затронуты: у них был обнаружен больший индекс массы тела.
Подбробнее про эпигенетику можно прочитать в нашем материале Эпигенетика — над ДНК.
Как ДНК передается детям?
У человека в ядре каждой клетки хранятся 23 пары хромосом, всего 46 штук. Исключение составляют половые клетки: сперматозоиды и яйцеклетки. В них содержится всего 23 одиноких хромосомы.
Число хромосом в половых клетках сокращается в результате мейоза, это особый тип деления клетки, в процессе которого из одной клетки с двумя наборами хромосом (диплоидной; 46 хромосом) образуется четыре с одинарным набором хромосом (гаплоидных; 23 хромосомы).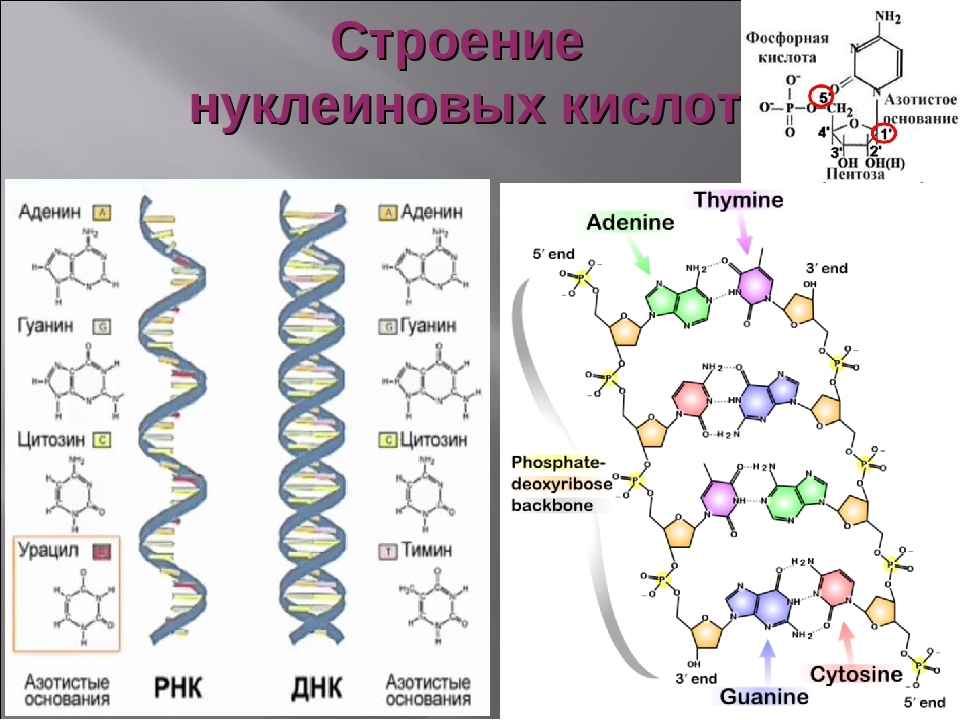 Хромосомы, содержащие одну и ту же последовательность генов и образующие пары, могут обмениваться участками — происходит кроссинговер, своеобразное перемешивание, которое обеспечивает потомкам большее генетическое разнообразие. Когда мужская и женская половые клетки сливаются при оплодотворении — в зародыше снова оказывается двойной набор генов, однако смешанный: по одной хромосоме в паре от папы и по одной — от мамы. Например, мы рассказывали, как от мамы и папы может наследоваться интеллект и как наследуются некоторые заболевания, например, галактоземия. Сокращение набора хромосом необходимо, чтобы, когда сперматозоид и яйцеклетка встретились при оплодотворении, образовался организм с нормальным набором хромосом. Если хромосом больше или меньше, чем нужно,- возникают хромосомные болезни. Классический пример — Болезнь Дауна, при которой вместо двух хромосом под номером 21 в организме оказывается три.
Хромосомы, содержащие одну и ту же последовательность генов и образующие пары, могут обмениваться участками — происходит кроссинговер, своеобразное перемешивание, которое обеспечивает потомкам большее генетическое разнообразие. Когда мужская и женская половые клетки сливаются при оплодотворении — в зародыше снова оказывается двойной набор генов, однако смешанный: по одной хромосоме в паре от папы и по одной — от мамы. Например, мы рассказывали, как от мамы и папы может наследоваться интеллект и как наследуются некоторые заболевания, например, галактоземия. Сокращение набора хромосом необходимо, чтобы, когда сперматозоид и яйцеклетка встретились при оплодотворении, образовался организм с нормальным набором хромосом. Если хромосом больше или меньше, чем нужно,- возникают хромосомные болезни. Классический пример — Болезнь Дауна, при которой вместо двух хромосом под номером 21 в организме оказывается три.
На заметку.
1. Инструкция о строении нашего тела закодирована в ДНК в формате последовательностей нуклеотидов А, Т, Г , Ц.
2. Длинная молекула ДНК компактно упаковывается в ядре клетки с помощью скручивания и наматывания на белки.
3. Молекула ДНК удваивается в процессе репликации.
4. Чтобы извлечь информацию из ДНК, ее надо перевести на РНК.
5. Получившуюся РНК читают органеллы рибосомы по триплетам из трех букв.Триплет соответствует аминокислоте — звену цепи белка. Поэтому читая триплеты, РНК понимают, какую аминокислоту следующей присоединить к растущей к цепи белка.
6. Белки определяют почти все наши свойства и функции.
7. Гены можно активировать и подавлять с помощью специальных белков, транскрипционных факторов и супрессоров, с помощью изменения гистонов — катушек, на которые намотана ДНК, или с помощью метилирования ДНК.
8. На работу генов, в том числе на их активацию и подавление, могут влиять внешние факторы: стресс, ночные смены, диета и так далее (однако последовательность нуклеотидов ДНК при этом не меняется).
9. В клетках нашего тела содержится 23 пары хромосом. Но в половых клетках хромосомы содержатся не парами, а одиночно (гаплоидный набор хромосом). Такое сокращение необходимо, чтобы при оплодотворении, образовался организм с нормальным набором хромосом.
Такое сокращение необходимо, чтобы при оплодотворении, образовался организм с нормальным набором хромосом.
Если вам интересны особенности собственной ДНК, то в результатах Генетического теста Атлас вы можете найти целый раздел о необычных признаках, которые определяют ваши вкусы, чувствительность к определенным запахам и привычки.
О том, как еда изменяет едоков | Научные открытия и технические новинки из Германии | DW
Обмен веществ в живом организме — процесс чрезвычайно сложный, особенно когда речь идет о высокоразвитых организмах. Поэтому столь важную роль играют механизмы, регулирующие этот процесс. Сравнительно недавно ученые обнаружили, что в регуляции обмена веществ участвуют не только ферменты, гормоны, факторы роста и тому подобные соединения, но и так называемые микроРНК. Вообще РНК, то есть рибонуклеиновые кислоты, выполняют в живых клетках множество самых разных функций: так, матричная РНК служит посредником в передаче наследственной информации, закодированной в ДНК, рибосомам, синтезирующим на основе этой информации белки, транспортная РНК доставляет аминокислоты к месту синтеза белков.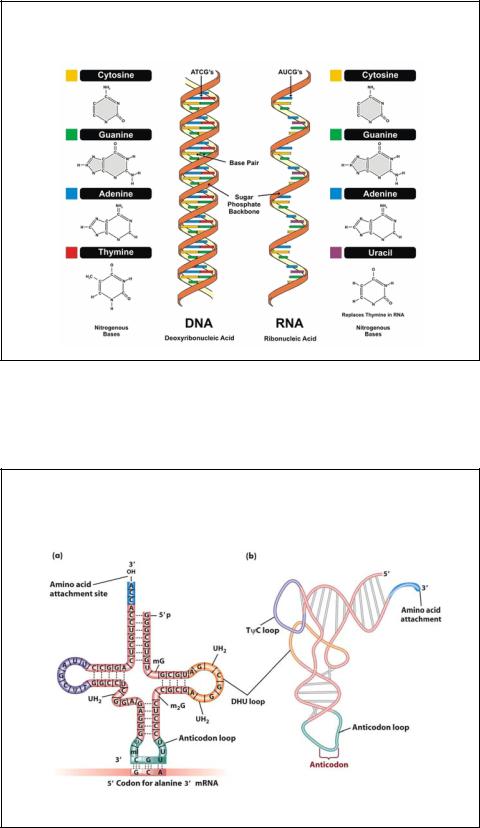 Есть и другие классы этих так называемых малых некодирующих РНК — они участвуют, например, в регуляции генов или играют роль биологических катализаторов химических реакций в организме.
Есть и другие классы этих так называемых малых некодирующих РНК — они участвуют, например, в регуляции генов или играют роль биологических катализаторов химических реакций в организме.
Седьмой класс питательных веществ
Так вот, о том, что микроРНК, то есть короткие, длиной всего в 2 десятка нуклеотидов, молекулы рибонуклеиновых кислот, способны замедлять или даже блокировать производство того или иного белка, ученые уже знали. Но то, что теперь опубликовала в авторитетном научном журнале Cell Research группа китайских исследователей, вызвало у специалистов немалое изумление. Профессор цитологии Чэнь-Ю Чжан (Chen-Yu Zhang) и его коллеги из Нанкинского университета обследовали 50 добровольцев и обнаружили в их крови и тканях… микроРНК растительного происхождения.
Это и само по себе стало изрядной неожиданностью, поскольку до сих пор считалось, что все растительные ДНК и РНК, попадающие в организм человека с пищей, полностью разлагаются, разрушаются в процессе переваривания. Но еще большее удивление вызвал тот факт, что эти растительные микроРНК участвуют в регуляции метаболизма человека наравне с его собственными микроРНК. По словам руководителя исследования, это открытие заставляет совершенно по-новому взглянуть на роль питания в жизни человека: «Считается, что существует шесть классов питательных веществ — белки, жиры, углеводы, витамины, минеральные вещества и вода. Однако теперь выясняется, что еще и растительные микроРНК, судя по всему, оказывают на активность наших генов, а значит, и на наш обмен веществ, самое непосредственное воздействие. Это дает основание считать их седьмым классом питательных веществ».
Но еще большее удивление вызвал тот факт, что эти растительные микроРНК участвуют в регуляции метаболизма человека наравне с его собственными микроРНК. По словам руководителя исследования, это открытие заставляет совершенно по-новому взглянуть на роль питания в жизни человека: «Считается, что существует шесть классов питательных веществ — белки, жиры, углеводы, витамины, минеральные вещества и вода. Однако теперь выясняется, что еще и растительные микроРНК, судя по всему, оказывают на активность наших генов, а значит, и на наш обмен веществ, самое непосредственное воздействие. Это дает основание считать их седьмым классом питательных веществ».
Рис как фактор, регулирующий расщепление холестерина
В частности, Чэнь-Ю Чжан обнаружил у всех обследованных добровольцев в плазме крови и клетках печени микроРНК типа MIR168a. Весьма обильно эти молекулы присутствуют в рисе. Опыты на трансгенных мышах показали, что в организме человека MIR168a блокирует синтез чрезвычайно важного белка — так называемого клеточного рецептора липопротеинов низкой плотности. Этот белок самым непосредственным образом связан с транспортировкой холестерина и его расщеплением в печени. Таким образом, потребление риса в пищу не только обеспечивает организм человека энергией, но и регулирует активность одного из важных генов, влияя тем самым на обмен веществ и на здоровье человека. Ведь повышенный уровень содержания в крови липопротеинов низкой плотности увеличивает риск атеросклероза.
Этот белок самым непосредственным образом связан с транспортировкой холестерина и его расщеплением в печени. Таким образом, потребление риса в пищу не только обеспечивает организм человека энергией, но и регулирует активность одного из важных генов, влияя тем самым на обмен веществ и на здоровье человека. Ведь повышенный уровень содержания в крови липопротеинов низкой плотности увеличивает риск атеросклероза.
Как растительные микроРНК умудряются уцелеть в пищеварительном тракте человека и проникнуть оттуда в кровь, пока неясно, признает Чэнь-Ю Чжан: «Нам неизвестен этот механизм в деталях. Однако мы полагаем, что эти растительные микроРНК могут захватываться клетками эндотелия сосудов кишечной стенки. При этом мембраны эндотелиальных клеток формируют особые внеклеточные структуры, в которые, как в оболочку, заключаются микроРНК. В таких миниатюрных пузырьках, называемых экзосомами, микроРНК поступают в кровоток».
Новые основы старой китайской медицины
По мнению ученого, его открытие позволяет по-новому объяснить лечебные свойства лекарственных трав, широко применяемых в традиционной китайской медицине. В ходе экспериментов, результаты которых еще только ждут опубликования, Чэнь-Ю Чжан подмешивал экстракт из растения, известный в Китае как эффективное средство против симптомов гриппа, в корм подопытным мышам, которых предварительно инфицировали вирусом инфлюэнцы. Вскоре микроРНК этого растения обнаружились в легочной ткани мышей, где они заблокировали синтез белка, необходимого для размножения вируса, и тем самым предотвратили заболевание. «Это открытие поистине революционно, — не без гордости говорит исследователь. — Возможно, мы начнем вскоре применять различные чужеродные микроРНК — и не только растительные, но и животные, — для лечения болезней. А вводить эти препараты в организм можно будет просто с пищей».
В ходе экспериментов, результаты которых еще только ждут опубликования, Чэнь-Ю Чжан подмешивал экстракт из растения, известный в Китае как эффективное средство против симптомов гриппа, в корм подопытным мышам, которых предварительно инфицировали вирусом инфлюэнцы. Вскоре микроРНК этого растения обнаружились в легочной ткани мышей, где они заблокировали синтез белка, необходимого для размножения вируса, и тем самым предотвратили заболевание. «Это открытие поистине революционно, — не без гордости говорит исследователь. — Возможно, мы начнем вскоре применять различные чужеродные микроРНК — и не только растительные, но и животные, — для лечения болезней. А вводить эти препараты в организм можно будет просто с пищей».
Собственно, идея использовать микроРНК в качестве биологически активного компонента лекарств обсуждается в фармацевтике уже давно. Но до сих пор все эксперименты упирались в одну неразрешимую проблему: как доставить микроРНК точно и целенаправленно в нужное место в организме. Исследования китайских цитологов показали, что природа уже давно предусмотрительно создала такие пути и что функция пищи очевидно не сводится к одному лишь обеспечению организма энергией.
Исследования китайских цитологов показали, что природа уже давно предусмотрительно создала такие пути и что функция пищи очевидно не сводится к одному лишь обеспечению организма энергией.
Автор: Владимир Фрадкин
Редактор: Марина Борисова
ДНК. Механизмы хранения и обработки информации. Часть I / Хабр
Много людей использует термин ДНК. Но статей, нормально описывающих, как она работает почти нет (понятных не биологам). Я уже описывал в общих чертах
устройство клеткии самые основы ее
энергетических процессов. Теперь перейдем к ДНК.
ДНК хранит информацию. Это знают все. Но вот как она это делает?
Начнем с того, где она в клетке хранится. Примерно 98% хранится в ядре. Остальное в митохондриях и хлоропластах (в этих ребятах протекает фотосинтез). ДНК — это огромный полимер, состоящий из мономерных звеньев. Выглядит примерно так.
Что мы тут видим? Во-первых ДНК — двухцепочечная молекула. Почему это так важно — чуть позже. Далее мы видим синие пятиугольники. Это молекулы дезоксирибозы (такой сахар, чуть меньше глюкозы. От рибозы отличается отсутствием одной OH группы, что придает стабильности молекуле ДНК, в отличие от РНК, в которой используется рибоза. Дальше, для простоты опущу приставку дезокси и буду просто говорить рибоза, да простят нас щепетильные товарищи). Маленькие кружкИ — остатки фосфорной кислоты. Ну и собственно есть азотистые основания. Всего их 5, но в ДНК в основном встречаются 4. Это Аденин, Гуанин, Тимин и Цитозин. То есть, есть рибоза с которой связано азотистое основание. Вместе они образуют так называемые нуклеозиды, которые связываются друг с другом с помощью остатков фосфорной кислоты. Таким образом мы получаем длинную цепь, состоящую из мономеров. Теперь посмотрите на увеличенную левую цепь. Видите C и G соединены тремя пунктирными линиями, а T и A двумя. Что это значит? Да, ДНК состоит из двух цепей, но что удерживает их вместе? Есть такая штука, как водородная связь. Выглядит примерно так.
Далее мы видим синие пятиугольники. Это молекулы дезоксирибозы (такой сахар, чуть меньше глюкозы. От рибозы отличается отсутствием одной OH группы, что придает стабильности молекуле ДНК, в отличие от РНК, в которой используется рибоза. Дальше, для простоты опущу приставку дезокси и буду просто говорить рибоза, да простят нас щепетильные товарищи). Маленькие кружкИ — остатки фосфорной кислоты. Ну и собственно есть азотистые основания. Всего их 5, но в ДНК в основном встречаются 4. Это Аденин, Гуанин, Тимин и Цитозин. То есть, есть рибоза с которой связано азотистое основание. Вместе они образуют так называемые нуклеозиды, которые связываются друг с другом с помощью остатков фосфорной кислоты. Таким образом мы получаем длинную цепь, состоящую из мономеров. Теперь посмотрите на увеличенную левую цепь. Видите C и G соединены тремя пунктирными линиями, а T и A двумя. Что это значит? Да, ДНК состоит из двух цепей, но что удерживает их вместе? Есть такая штука, как водородная связь. Выглядит примерно так. На атомы кислорода (O) и азота (N) формируется частичный отрицательный заряд, а на водороде (H) — положительный. Это приводит к формированию слабых связей.
На атомы кислорода (O) и азота (N) формируется частичный отрицательный заряд, а на водороде (H) — положительный. Это приводит к формированию слабых связей.
Связи действительно очень слабые. Их энергия может быть в 200 раз ниже энергии ковалентных связей (образуются за счет перекрытия пары электронных облаков, например связь в молекуле CO2). Однако таких связей много. В каждой нашей клетке ДНК цепи связаны почти 16 миллиардами слабых связей, не мало, согласны?
Но вернемся к числу связей между основаниями. Цитозин и Гуанин связаны тремя связями, а Аденин и Тимин — двумя. Это приводит к тому, что Г и Ц связанны куда прочнее, чем А и Т. Некоторым организмам нужна особая стабильность связей ДНК, например живущим при высоких температурах. При нагревании ДНК содержащая больше ГЦ пар более стабильна. Так что хочешь жить в гейзере — имей много ГЦ пар. Хотя последние исследования говорят, что явной связи между GC составом (% ГЦ пар от всех пар) и температурой обитания нет. Стоит сказать, что варьирует он сильно. Так у Candidatus Carsonella ruddii PV (внутриклеточный эндосимбионт) он примерно 16%, у нас с вами почти 41%, а у Anaeromyxobacter K (бактерия вполне себе средних размеров) достигает 75%.
Стоит сказать, что варьирует он сильно. Так у Candidatus Carsonella ruddii PV (внутриклеточный эндосимбионт) он примерно 16%, у нас с вами почти 41%, а у Anaeromyxobacter K (бактерия вполне себе средних размеров) достигает 75%.
Тут вы можете видеть связь GC состава с размером генома бактерий. Mb — миллион пар нуклеотидов. Показатель довольно вариативный. Его, кстати, часто юзают как фичу при обучении различного рода классификаторов. Сам недавно писал классификатор для распознания патогенов на основе сырых данных секвенирования и оказалось, что GC состав даже по одному риду вполне себе можно использовать.
Пока не забыл. Почему важно, что ДНК двухцепочечная? На основе одной цепи можно восстановить другую. Если в одной цепи поврежден кусок напротив последовательности Аденин-Аденин-Цитозин, то мы точно знаем, что до повреждения там был Тимин-Тимин-Гуанин. Таким образом наличие второй цепи позволяет надежней хранить информацию.
Круто! Теперь вернемся к самой молекуле ДНК. Это цепочка из 4х типов звеньев. Однако насколько длинная? У Candidatus Carsonella ruddii PV уже упомянутого выше всего 160 000 нуклеотидов. У нас с вами 3.2 миллиарда (в гаплоидной клетке, то есть с одним набором хромосом. У большинства наших клеток их два). Кажется много, да? На самом деле нет. У одноклеточной амебы (Amoeba dubia) он примерно 670 миллиардов пар нуклеотидов. Кажется что это бесконечно длинная цепочка, поэтому давайте переведем размер в любимые нам метры. Если все наши хромосомы (их 46, не забываем; 23 по две копии на каждую) развернуть и вытянуть в одну линию, получится примерно 2х метровая цепочка. ДНК одной амебы хватит, чтоб опоясать футбольный стадион. Но к чему я веду? Ядро, в котором ДНК хранится не очень большое. У нас оно в среднем диаметром в 6 мкм. Не очень то много, если хочешь свернуть 2х метровую нить, пусть и очень тонкую. Причем нужно не просто запихать нить в ядро. Нужно свернуть таким образом, чтобы в любой момент можно было обеспечить доступ к любому ее участку.
Это цепочка из 4х типов звеньев. Однако насколько длинная? У Candidatus Carsonella ruddii PV уже упомянутого выше всего 160 000 нуклеотидов. У нас с вами 3.2 миллиарда (в гаплоидной клетке, то есть с одним набором хромосом. У большинства наших клеток их два). Кажется много, да? На самом деле нет. У одноклеточной амебы (Amoeba dubia) он примерно 670 миллиардов пар нуклеотидов. Кажется что это бесконечно длинная цепочка, поэтому давайте переведем размер в любимые нам метры. Если все наши хромосомы (их 46, не забываем; 23 по две копии на каждую) развернуть и вытянуть в одну линию, получится примерно 2х метровая цепочка. ДНК одной амебы хватит, чтоб опоясать футбольный стадион. Но к чему я веду? Ядро, в котором ДНК хранится не очень большое. У нас оно в среднем диаметром в 6 мкм. Не очень то много, если хочешь свернуть 2х метровую нить, пусть и очень тонкую. Причем нужно не просто запихать нить в ядро. Нужно свернуть таким образом, чтобы в любой момент можно было обеспечить доступ к любому ее участку.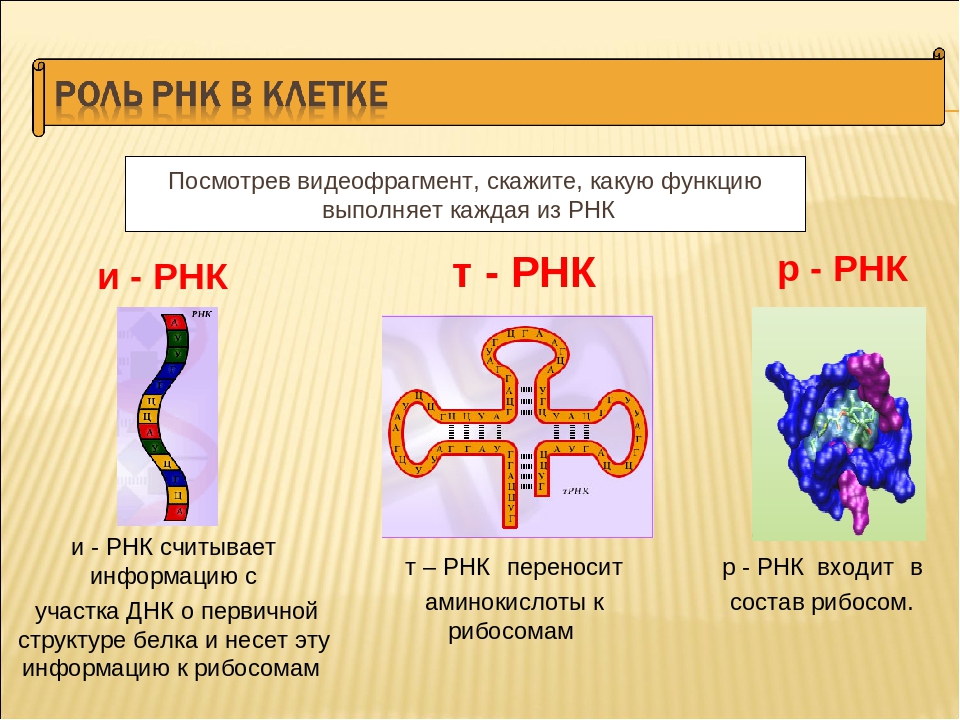 Задача сложная. И с ней успешно справляются специализированные белки. Они создают ряд спиралей и петель, которые обеспечивают все более и более высокие уровни упаковки и не до допускают спутывания ДНК в гордиев узел. Давайте поговорим о том, как она упаковывается.
Задача сложная. И с ней успешно справляются специализированные белки. Они создают ряд спиралей и петель, которые обеспечивают все более и более высокие уровни упаковки и не до допускают спутывания ДНК в гордиев узел. Давайте поговорим о том, как она упаковывается.
Сразу скажу, упаковывается она очень по разному. Но если откинуть экзотику, то остается два способа. Первый характерен для бактерий, второй для эукариот (или иначе ядерных).
Упаковка ДНК у бактерий
Начнем с братьев наших меньших. Бактерии сами по себе обладают не очень большим геномом, в среднем от 1 до 5 миллионов пар нуклеотидов. Наиболее характерное их отличия от нас в том, что у них нет ядра и ДНК плавает в клетке. Не совсем плавает, оно частично прикреплено к клеточной мембране и тоже свернуто, но не так сильно как у нас.
Второе. Бактериальная ДНК чаще всего кольцевая. Так ее проще копировать (нет концов, которые могут потеряться при копировании и не нужно придумывать механизмы сохранения концов).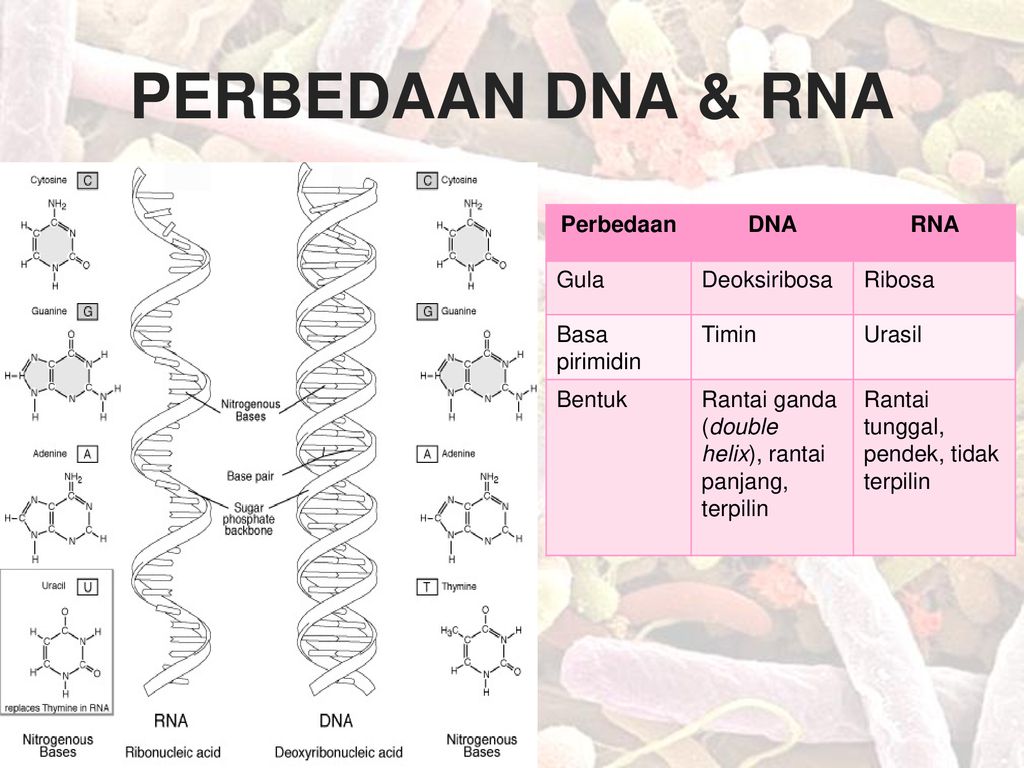 Обычно такое кольцо одно, но у некоторых бактерий их может быть 2 или 3. Есть еще кольца поменьше (от пары тысяч до пары сотен тысяч остатков).Имя им плазмиды, и это вообще отдельная история.
Обычно такое кольцо одно, но у некоторых бактерий их может быть 2 или 3. Есть еще кольца поменьше (от пары тысяч до пары сотен тысяч остатков).Имя им плазмиды, и это вообще отдельная история.
Вернемся к упаковке ДНК. ДНК упаковывают белки-гистоны (есть еще гистоноподобные белки). ДНК это дезоксирибонуклеиновая кислота. Кислота. Это значит что она отрицательно заряжена (за счет остатков фосфорной кислоты). Поэтому белки, связывающие ее положительно заряжены. Таким образом они могут связываются с ДНК. ДНК бактерий вместе с белками ее упаковывающими формируют нуклеоид, при этом на долю ДНК приходится 80% от его массы. Выглядит это примерно так. То есть кольцевая ДНК делится на домены по 40 тысяч пар нуклеотидов. Затем происходит скручивание. Внутри доменов тоже происходит скручивания, но его степень в разных доменах отличается. В среднем степень упаковки бактериальной ДНК варьирует от сотни до тысячи раз.
Есть еще прикольное видео.
Упаковка ДНК у эукариот
Тут все куда интересней. Наше ДНК хорошо упакована и спрятана внутри ядра. И она куда эффективней упакована, нежели у бактерий. Во время митоза (деление клетки) размер 22й хромосомы составляет 2 мкм. Если ее распутать и вытянуть, она будет уже 1,5 см. Что соответствует степени упаковки в 10 000 раз. Это около максимальная степень упаковки нашей ДНК. Во время деления нужно максимально упаковать ДНК, что бы эффективно разделить ее между дочерними клетками. В обыденной жизни степень компактизации составляет примерно 500 раз. Со слишком упакованной ДНК сложно считывать информацию.
Наше ДНК хорошо упакована и спрятана внутри ядра. И она куда эффективней упакована, нежели у бактерий. Во время митоза (деление клетки) размер 22й хромосомы составляет 2 мкм. Если ее распутать и вытянуть, она будет уже 1,5 см. Что соответствует степени упаковки в 10 000 раз. Это около максимальная степень упаковки нашей ДНК. Во время деления нужно максимально упаковать ДНК, что бы эффективно разделить ее между дочерними клетками. В обыденной жизни степень компактизации составляет примерно 500 раз. Со слишком упакованной ДНК сложно считывать информацию.
Есть несколько уровней упаковки ДНК эукариот
Первый — нуклеосомный уровень. 8 белков-гистонов формируют частицу на которую наматывается ДНК. Затем еще один белок ее фиксирует. Выглядит примерно так.
Получаются своего рода бусы. Плотность упаковки благодаря этому возрастает в 7-10 раз. Далее нуклеосомы упаковываются в фибрилы. Немного похоже на солениод. Тут суммарная степень упаковки может достигать 60 раз.
Следующий этап компактизации ДНК связан с образованием петлеобразных структур, которые называются хромомерами. Фибрила разбита на участки по 10 — 80 тысяч пар азотистых оснований. В местах разбивки находятся глобулы негистоновых белков. ДНК — связывающие белки узнают глобулы негистоновых белков и сближают их. Образуется устье петли. Средняя длина петли включает примерно 50 тысяч оснований. Эту структуру называют интерфазной хромонемой. И именно в ней наше ДНК находится большую часть времени. Уровень упаковки здесь достигает 500-1500 раз.
При необходимости клетка может еще больше компактизировать генетический материал. Идет образование более крупных петель из хромомерной фибриллы. Эти петли в свою очередь образуют новые петли (петли в петли… и это не вязание). Которые в конечном счете формируют хромосому.
В целом процесс упаковки можно описать примерно так.
В итоге из нитей ДНК мы получаем, при делении, суперскрученные структуры, которые можно увидеть под микроскопом. Их мы и зовем хромосомами.
Их мы и зовем хромосомами.
Собственно вещество хромосом зовется хроматином. И степень его упаковки отличается в зависимости от участка хромосомы. Есть эухроматин и гетерохроматин. Эухроматин это довольно расплетенная область хроматина, в ней ДНК находится на хромомерном уровне (упаковка в 500 — 1000 раз). Здесь происходит активное считывание информации. Например, если сейчас клетка активно синтезирует белок А, то область ДНК, его кодирующая будет в состоянии эухроматина, что бы ферменты, «читающие» ДНК могли до нее добраться. Гетерохроматин же содержит ту часть ДНК, которая клетке не особо нужна сейчас. То есть ДНК максимально плотно упакована, дабы не путаться под ногами. В зависимости от потребностей клетки одни области хроматина могут частично расплетаться, в то время как другие — сплетаться. Таким образом еще и осуществляется регуляция (очень грубое приближение), ведь к скрученной области не добраться, и значит ее не прочитать.
Собственно пока это все. Мы обсудили как хранится носитель информации.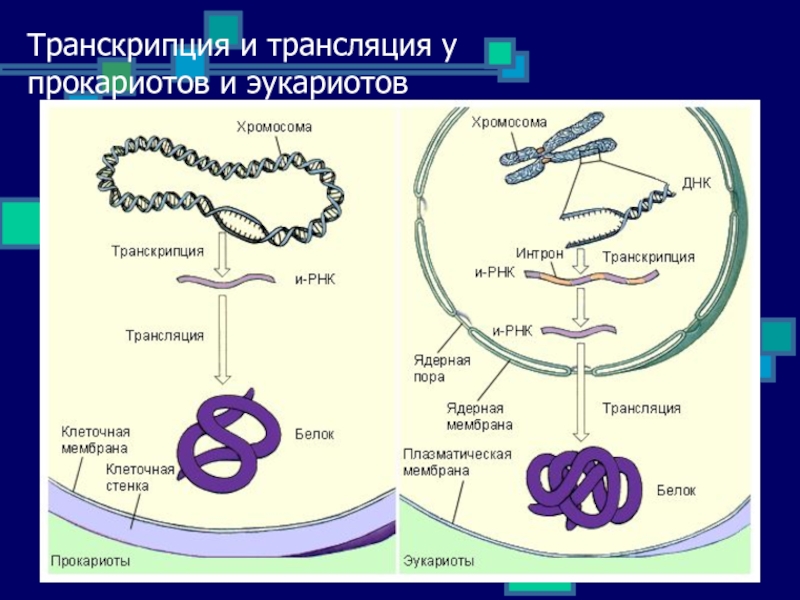 Сделаем небольшую паузу и через пару дней поговорим о самом кодировании информации.
Сделаем небольшую паузу и через пару дней поговорим о самом кодировании информации.
clonealign: статистическая интеграция независимых данных секвенирования РНК и ДНК одной клетки рака человека | Биология генома
Clonealign: построение модели и вывод
Начнем с матрицы экспрессии N × G необработанных прочтений Y для N клеток и 6 G генов 900 и 6 G 900 и G 900 × C матрица Λ = ( λ gc ) клоноспецифических номеров копий для клонов C и генов G .Такую матрицу числа копий обычно получают путем филогенетического анализа данных одноклеточной CNV с последующим разрезанием филогенетического дерева для получения C клонов или клад. Цель вывода состоит в том, чтобы отнести каждую из клеток N , измеренных в пространстве РНК, к одному из клонов C , измеренных в пространстве ДНК.
Для каждой ячейки n =1,…, N мы вводим категориальную переменную присвоения z n , определенную таким образом, что
$$ z_{n} = c\, \text{ если ячейка}\ n \text{ назначена для клонирования } c $$
(1)
для c =1,…, C . Наше предположение состоит в том, что y ng — экспрессия гена g в клетке n — будет зависеть от числа копий гена в клоне, которому присвоен n , т. е. \ (\mathbb {E} \propto \mu _{g} f(\lambda _{gc})\) где μ g — экспрессия гена на одну копию g и f — дозировочная функция , которая сопоставляет количество копий гена с мультипликативным фактором экспрессии.Хотя эта функция априори неизвестна, и совместная оценка с клональными популяциями привела бы к неидентифицируемой модели, мы можем закодировать некоторые основные предположения о дозировке генов в нашу спецификацию f . Мы предполагаем, что если изменение числа копий небольшое, это приведет к пропорциональному изменению выражения, например, число копий, равное 3, предположительно может привести к \(\frac {3}{2}\times \) большему выражению. И наоборот, мы предполагаем, что если изменение числа копий велико, например, если клон имеет число копий 12 в определенной области, клетки будут иметь компенсаторный механизм, такой, что меньше, чем \(\frac {12}{2} \times \) производятся стенограммы, и это ограничено верхним пределом.
Наше предположение состоит в том, что y ng — экспрессия гена g в клетке n — будет зависеть от числа копий гена в клоне, которому присвоен n , т. е. \ (\mathbb {E} \propto \mu _{g} f(\lambda _{gc})\) где μ g — экспрессия гена на одну копию g и f — дозировочная функция , которая сопоставляет количество копий гена с мультипликативным фактором экспрессии.Хотя эта функция априори неизвестна, и совместная оценка с клональными популяциями привела бы к неидентифицируемой модели, мы можем закодировать некоторые основные предположения о дозировке генов в нашу спецификацию f . Мы предполагаем, что если изменение числа копий небольшое, это приведет к пропорциональному изменению выражения, например, число копий, равное 3, предположительно может привести к \(\frac {3}{2}\times \) большему выражению. И наоборот, мы предполагаем, что если изменение числа копий велико, например, если клон имеет число копий 12 в определенной области, клетки будут иметь компенсаторный механизм, такой, что меньше, чем \(\frac {12}{2} \times \) производятся стенограммы, и это ограничено верхним пределом. С учетом этих соображений мы определяем f как \text {if } \lambda \geq \zeta, \end {массив}\right.\)где в нашем анализе мы фиксируем ζ =6. Мы оставляем на будущее более сложные подходы, такие как вывод f из совместных геномно-транскриптомных анализов или маргинализация ζ в байесовских моделях.
С учетом этих соображений мы определяем f как \text {if } \lambda \geq \zeta, \end {массив}\right.\)где в нашем анализе мы фиксируем ζ =6. Мы оставляем на будущее более сложные подходы, такие как вывод f из совместных геномно-транскриптомных анализов или маргинализация ζ в байесовских моделях.
Затем мы указываем точную модель правдоподобия для clonealign.В моделировании данных RNA-seq, как описано в [31], есть одна тонкость, заключающаяся в том, что экспрессия каждого гена измеряется относительно всех других генов в данной библиотеке, умноженной на глубину секвенирования этой библиотеки. Принятие этого во внимание имеет решающее значение для нашей проблемы, поскольку, если ген с высокой экспрессией находится в области с большим числом копий в клоне, это вызовет снижение экспрессии всех других генов. Таким образом, ожидаемое количество гена g в ячейке n при условии, что эта клетка отнесена к клону c , определяется как:
$$ \begin{aligned} &\mathbb{E}[y_{ng}|z_{n}=c] =\\ &\underbrace{s_{n}}_{\substack{\text{Чтение ячейки } \\ \text{глубина}}} \frac{\overbrace{\mu_{g}}^{\substack{\text{Per-copy} \\ \text{выражение}}} \times \text{ \; } f (\ overbrace {\ lambda_ {gc}} ^ {\ substack {\ text {Копировать} \\ \ text {число}}}) \ times e ^ {\ overbrace {\ boldsymbol {x} _ {n} \ cdot \boldsymbol{\beta}_{g}^{T}}^{\substack{\text{известные} \\ \text{ковариаты}}} + \overbrace{\boldsymbol{\psi}_{n} \ cdot \boldsymbol{w}_{g}^{T}}^{\substack{\text{Остаток} \\ \text{выражение}}}}} {\underbrace{\sum\limits_{g’=1} ^{G} \mu_{g’} f(\lambda_{g’c}) e^{\boldsymbol{x}_{n} \cdot \boldsymbol{\beta}_{g’}^{T} + \boldsymbol{\psi}_{n} \cdot \boldsymbol{w}_{g’}^{T}}}_{\text{Общая нормализация РНК}}} \end{выровнено} $$
(2)
где s n — это общий размер глубины чтения ячейки n . {T} \) между рядными векторами ψ N 2 N Q N × Q × ψ 2 и векторы ряда W 2 G G × Q матрица W вводит в модель структурированный шум и позволяет избежать принудительного объяснения всех изменений выражения в терминах изменения количества копий, что на практике неверно. Этот термин аналогичен представлению наблюдаемых данных как произведения двух матриц низкого ранга в таких моделях, как факторный анализ и линейные модели смешанных эффектов.{T}\) влияет на среднее значение аналогично случайным эффектам, как указано выше. Ковариаты x n могут кодировать известные группы клеток, такие как эффекты экспериментальных партий, или дополнительную биологическую информацию, такую как стадия клеточного цикла, которую можно вывести экспериментально или из данных об экспрессии генов с использованием таких методов, как сканирование [32].
{T} \) между рядными векторами ψ N 2 N Q N × Q × ψ 2 и векторы ряда W 2 G G × Q матрица W вводит в модель структурированный шум и позволяет избежать принудительного объяснения всех изменений выражения в терминах изменения количества копий, что на практике неверно. Этот термин аналогичен представлению наблюдаемых данных как произведения двух матриц низкого ранга в таких моделях, как факторный анализ и линейные модели смешанных эффектов.{T}\) влияет на среднее значение аналогично случайным эффектам, как указано выше. Ковариаты x n могут кодировать известные группы клеток, такие как эффекты экспериментальных партий, или дополнительную биологическую информацию, такую как стадия клеточного цикла, которую можно вывести экспериментально или из данных об экспрессии генов с использованием таких методов, как сканирование [32].
Мы вводим отрицательное биномиальное правдоподобие, которое обычно используется для моделирования данных секвенирования РНК [31, 33] и одноклеточной секвенирования РНК [34] со средним значением, заданным уравнением.{2}) $$
(3)
, где местоположение c i и с b каждой базисной функции является фиксированным гиперпараметром, а амплитуда каждого a i выводится совместно из данных. Мы фиксируем M = 20 по умолчанию и равномерно распределяем c i от минимального до максимального исходных значений счета и устанавливаем b = 1/(2 δ 2 ), где
5 6 расстояние между последовательными основаниями.
Модель, определенная в 2, инвариантна к перемасштабированию всех µ , поэтому мы фиксируем µ 1 =1 и интерпретируем оставшиеся µ 2 , 05 90 90 ,…, µ — выражение для каждой копии относительно гена 1 с предшествующим \(\log \mu _{g} \sim \mathcal {N}(0,1)\).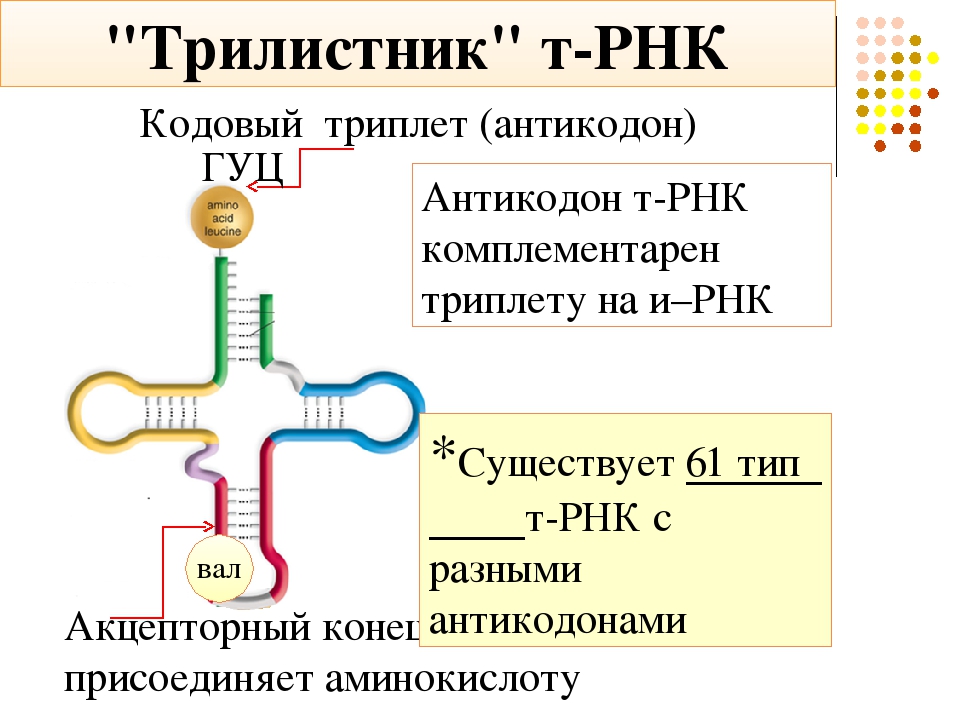 Общая глубина считывания s n может быть либо выведена совместно с моделью, либо зафиксирована заранее.
Общая глубина считывания s n может быть либо выведена совместно с моделью, либо зафиксирована заранее.
Вывод выполняется с использованием вариационного Байеса среднего поля (см., например,г., [36]). Кратко, учитывая совместное распределение P ( x , θ ) данных x и параметры модели θ 2, мы стремимся найти вариационное распределение Q ( θ | | ζ ), где ζ являются вариационными параметрами, которые приближаются к позами р ( θ | x ) путем минимизации KL [ Q ( θ | ζ )|| p ( θ | x )], расхождение Кульбаха-Лейблера между вариационным и апостериорным распределениями, что эквивалентно минимизации нижней границы доказательства (ELBO).Несопряженный характер модели в уравнении. 2 требует, чтобы мы вычислили оценку расхождения KL по методу Монте-Карло, которую мы можем оптимизировать, вычисляя градиенты с низкой дисперсией, используя прием репараметризации [37].
В частности, мы устанавливаем аппроксимирующее распределение вида \(q(\boldsymbol {z}, \boldsymbol {\mu }) = \prod _{n} q(z_{n}) \prod _{g} q (\mu _{g})\) для переменных присвоения клона и среднего выражения соответственно и оптимизировать все остальные параметры модели как вариационные параметры аналогично [37].Приближенное распределение для назначений клонов имеет категориальный вид q ( z n = c ) = φ 5 nc
6 . Аппроксимирующее распределение для параметров среднего выражения задается непрерывно дифференцируемым обратимым преобразованием стандартного гауссовского шума \(\epsilon \sim \mathcal {N}(0,1)\) на μ g = exp( ν г + ρ г ε ).В то время как математическое ожидание по q ( z ) может быть взято аналитически, чтобы вычислить математическое ожидание относительно q ( µ ), мы должны вычислить оценку Монте-Карло, взяв выборки S
6
µ ( s ) ∼ q ( µ ), где мы полагаем S =1, следуя предыдущей литературе [37].
Оптимизация выполняется с помощью оптимизатора Adam [38], реализованного в Tensorflow.Схождение оценивается путем мониторинга ELBO с моделью, сходящейся, когда изменение между последовательными итерациями падает ниже 10 −6 % . clonealign имеет открытый исходный код и доступен в Интернете по адресу http://www.github.com/kieranrcampbell/clonealign.
Включение информации об аллельном дисбалансе
Мы можем использовать информацию об аллельном дисбалансе в данных scRNA-seq для дальнейшего уточнения назначения клонотипа. Для экспрессированных SNP гетерозиготной зародышевой линии в областях с клон-специфическим числом копий, если имеет место клон-специфическое событие LOH, то аллельные отношения будут смещены в сторону 0 (потеря альт) или 1 (потеря ref) по сравнению с диплоидными областями, где соотношение аллелей должно быть сосредоточено вокруг \(\ frac {1}{2}\).Обратите внимание, что мы предполагаем (i) scDNA-seq слишком поверхностна для фазовых вариантов и (ii) не существует нейтрального по отношению к копиям LOH.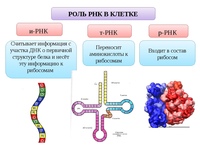 Если пользователь считает, что предположение (ii) нарушается при проверке прочтений scDNA-seq, тогда следует запустить clonealign, используя только данные об экспрессии генов.
Если пользователь считает, что предположение (ii) нарушается при проверке прочтений scDNA-seq, тогда следует запустить clonealign, используя только данные об экспрессии генов.
Мы определяем дополненную статистическую модель следующим образом: Пусть A NV R NV NV NV BE Alt и Ref Counts для (гетерозигозной зарок) Variant V в ячейке C для n =1,…, N и v =1,…, V .Кроме того, пусть λ vc будет номером копии варианта v в клоне c , полученным из данных scDNA-seq. Тогда вероятность, обусловленная клоном, определяется как:
$$ p(a_{nv}, r_{nv}) = \left\{ \begin{array}{ll} D_{\text{LOH}}(a_{nv}, r_{nv}) & \ text{if} \lambda_{vc} = 1 \\ D_{\text{HET}}(a_{nv}, r_{nv}) & \text{if} \lambda_{vc} = 2 \end{array} \правильно. $$
(4)
где
$$ \begin{align} D_{\text{HET}}(a_{nv}, r_{nv}) & = \text{BetaBinomial}(a_{nv}, a_{nv} + r_{nv} | \alpha = 2, \beta = 2) \\ D_{\text{LOH}}(a_{nv}, r_{nv}) & = \frac{1}{2}\text{BetaBinomial}(a_{ nv}, a_{nv} + r_{nv}|\alpha = 0. 1,\beta = 1,9) \\ & + \frac{1}{2} \text{BetaBinomial}(a_{nv}, a_{nv} + r_{nv} | \alpha = 1,9, \beta = 0,1) \end{выровнено} $$
1,\beta = 1,9) \\ & + \frac{1}{2} \text{BetaBinomial}(a_{nv}, a_{nv} + r_{nv} | \alpha = 1,9, \beta = 0,1) \end{выровнено} $$
(5)
Использование бета-биномиальной модели мотивировано наблюдением, что подсчеты прочтений будут следовать биномиальному распределению, но точное количество успешных прочтений (доля альтернативных прочтений) точно неизвестно из-за ошибок секвенирования и редактирования РНК, поэтому мы маргинализируем более это, чтобы получить данную модель наблюдения.Распределение D HET помещает массу вокруг фракции альтернативного аллеля \(\frac {1}{2}\), в то время как D LOH помещает его массу на 0 и 1. Калибровка дисперсии приводит к точному выбор параметров взят из недавнего исследования экспрессии клон-специфических аллелей в scRNAseq [39]. Вероятность, индуцированная уравнением 5 затем умножается на iid и добавляется к логарифмической объединенной вероятности данных и параметров для вариационного вывода, когда доступны данные SNV.-(Nukleinovye-kisloty)-otvet-na-vopros-3.jpg) Докеризированный рабочий процесс для создания требуемого варианта по клону и варианта по клеточным матрицам из выходных данных программного обеспечения 10X CellRanger и HMMCopy [15] соответственно доступен на http://www.github.com/kieranrcampbell/snvworkflow.
Докеризированный рабочий процесс для создания требуемого варианта по клону и варианта по клеточным матрицам из выходных данных программного обеспечения 10X CellRanger и HMMCopy [15] соответственно доступен на http://www.github.com/kieranrcampbell/snvworkflow.
Симуляции
Чтобы все симуляции были максимально реалистичными, модель clonealign была адаптирована к набору данных SA501, что дало эмпирическое распределение параметров модели и данных Λ ) р ( с ).Затем мы смоделировали на основе модели clonealign, выбрав выборку из эмпирического распределения параметров модели. Для clonealign мы рассмотрели пять различных сценариев моделирования, где каждый сценарий представляет собой предельный эффект, поскольку полная комбинация эффектов была бы неосуществимой с точки зрения вычислений. Во всех симуляциях сообщалось о площади под кривой оператора приемника (AUC) в качестве меры точности, за исключением изменения количества клонов, где мы используем точность в качестве метрики (доля клонов, называемых правильными).
Различное соотношение генов с эффектом дозировки
Для каждой симуляции определенная доля π =0,1,0,2,0,3,…,0,9 генов моделировалась с зависимостью от CN-экспрессии, в то время как экспрессия оставшейся доли 1− π имела выражение, не зависящее от число копий, достигаемое путем установки числа копий на 2 для всех клонов во время моделирования экспрессии, но предоставления истинного числа копий во время вывода, поскольку clonealign априори не знает, какие гены проявляют зависимость экспрессии CN.Наборы данных моделировали для двух клонов, соответствующих клонам А и В из SA501.
Различия в том, насколько геномно отличаются клоны
Количество генов, различающих клоны, в клон-специфических областях количества копий варьировалось от 2, 5, 10, 50, 100, 500 и 1000 на 1000 клеток и 2 клона.
Изменение количества клонов
Количество моделируемых клонов было установлено равным 2, 4, 8, 16, 32 и 64 для 200 и 800 генов и 1000 клеток.
Изменение частоты второстепенных клонов
Частота минорных клонов варьировала между 1 % , 5 % , 10 % , 20 % и 50 % для 200 и 800 генов и 1000 клеток.
Изменение качества данных scRNA-seq
Мы разделили смоделированные данные 10X от исходного размера набора данных 0,86 прочтений на ген на клетку до 1 % , 5 % , 10 % и 50 % для 200 и 800 генов и 1000 клеток.
Биоинформатический анализ
Для экспрессии всех данных scRNA-seq оценки были получены из необработанных подсчетов прочтений с использованием CellRanger (версия 2.0.1 для SA501X2B и версия 2.1.0 для (T)OV2295R), выровненных по hg19. При контроле качества клеток SA501X2B были удалены клетки с менее чем 1000 отсчетов или 350 экспрессированных генов в областях с различным числом копий между клонами A, B и C. Запросы числа копий, специфичные для клона, были созданы в соответствии с [1].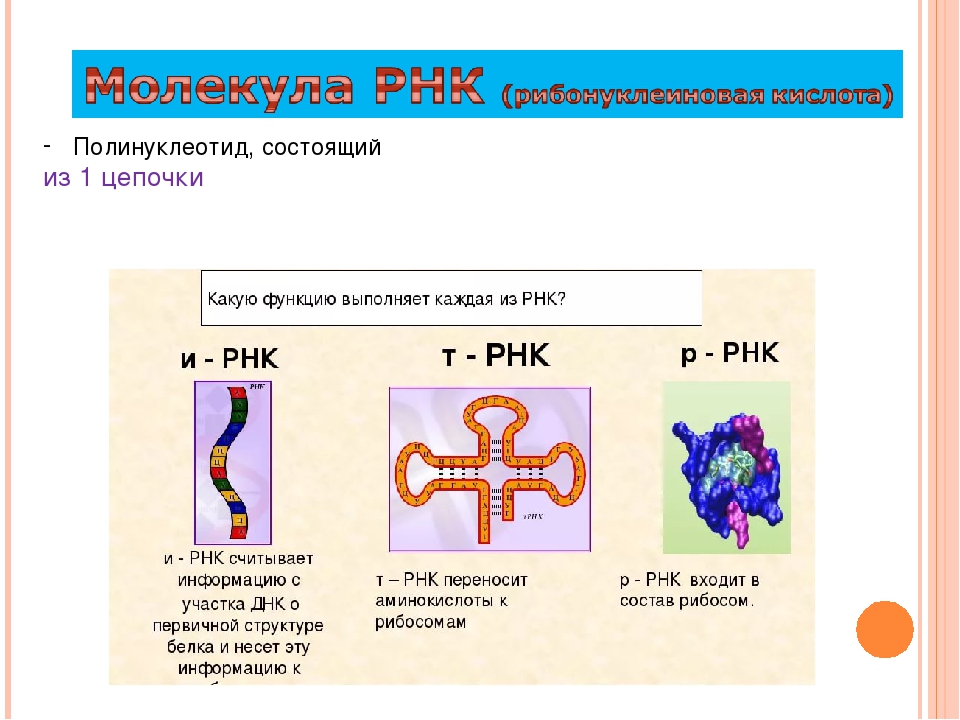 Гены Х-хромосомы были удалены до анализа выравнивания клонов, поскольку предположение о количестве копий экспрессии будет нарушено, если удаленная/амплифицированная копия Х неактивна.Для OV2295R и TOV2295R были сохранены клетки с общим UMI более 20 000 и общим количеством обнаруженных генов от 3000 до 7500. Вызовы числа копий для scDNA-seq были выполнены с использованием HMMCopy версии 1.22.0 и филогении, построенной с использованием модели скрытого дерева. . Число копий, специфичное для клона, было построено как среднее число копий всех клеток в клоне в данном положении. Гены на Х-хромосоме, как и прежде, были удалены.
Гены Х-хромосомы были удалены до анализа выравнивания клонов, поскольку предположение о количестве копий экспрессии будет нарушено, если удаленная/амплифицированная копия Х неактивна.Для OV2295R и TOV2295R были сохранены клетки с общим UMI более 20 000 и общим количеством обнаруженных генов от 3000 до 7500. Вызовы числа копий для scDNA-seq были выполнены с использованием HMMCopy версии 1.22.0 и филогении, построенной с использованием модели скрытого дерева. . Число копий, специфичное для клона, было построено как среднее число копий всех клеток в клоне в данном положении. Гены на Х-хромосоме, как и прежде, были удалены.
Анализ дифференциальной экспрессии (DE) проводили с использованием Limma Voom [16], версия 3.36.0. Для SA501X2B гены с более чем 100 общим количеством генов были сохранены для DE. Как для OV2295R, так и для TOV2295R гены с более чем 500 общими подсчетами были сохранены для DE, поскольку до этого порога соотношение средней дисперсии, о котором сообщила Лимма Вум, визуально плохо подходило. Все значения p были скорректированы для множественной проверки гипотез с использованием процедуры Бенджамини-Хохберга.
Все значения p были скорректированы для множественной проверки гипотез с использованием процедуры Бенджамини-Хохберга.
Для анализа SA501 LOH объемное полногеномное секвенирование ДНК, как описано ранее в [14], выравнивали по hg19 с использованием BWA aln версии 0.7.10, после чего аллели LOH зародышевой линии были идентифицированы с помощью samtools 1.7 mpileup с последующей командой VarScan 2.3.9 [40] mpileup2snp (настройки по умолчанию). Профили одноклеточной РНК и ДНК-seq были объединены в псевдообъемные клоны с использованием samtools версии 1.7 и картирования считываний с аллелями ref и alt в позициях, идентифицированных как гетерозиготные зародышевые линии, вызванные с помощью команды Varscan mpileup2cns с настройками по умолчанию, отличными от настройки –min-avg-qual 5. на слитой scRNA-seq, чтобы увеличить количество вызываемых позиций.Области в псевдообъемных скоплениях были названы LOH с использованием версии Titan 1.16.0 [41]. Мы сравнили частоту основного аллеля в области хромосомы 18, начиная с позиции 5,5×10 7 и далее, обнаружив значительно сниженную частоту основного аллеля в клоне А как в ДНК ( p =3,7×10 −51 ), так и в ДНК. РНК ( p = 5,9 × 10 -4 ), оба с использованием одностороннего критерия суммы рангов Уилкоксона.
РНК ( p = 5,9 × 10 -4 ), оба с использованием одностороннего критерия суммы рангов Уилкоксона.
Результаты моделирования на рис. 1d показывают, что чем выше латентная доля генов, которые проявляют зависимость от CN-гена, тем точнее наш вывод.Хотя набор генов, которые проявляют такую зависимость, априори неизвестен и, скорее всего, специфичен для рака и даже пациента, можно выбрать набор генов, которые с большей вероятностью будут проявлять такие взаимодействия, на основе предыдущих исследований. Например, мы взяли данные о количестве копий и экспрессии из когорт BRCA и OV из Атласа генома рака (TCGA, [42]) и регрессировали логарифмическое выражение на log R (относительное количество копий). Мы обнаружили, что подавляющее большинство генов демонстрируют положительную корреляцию с log R (дополнительный файл 2: рисунки S21 и S22).Только эти гены можно использовать в таких анализах, как клонирование.
Чтобы проверить устойчивость clonealign к выбору входных генов для наборов данных SA501, TOV2295R и OV2295R, мы повторно подогнали clonealign, за исключением нижних p % наименее вариабельных генов (как определено в логарифмическом пространстве экспрессии), для p ∈ {10, 20, 40, 60, 80, 90} и сравнили соответствие в назначении клонов между подходами.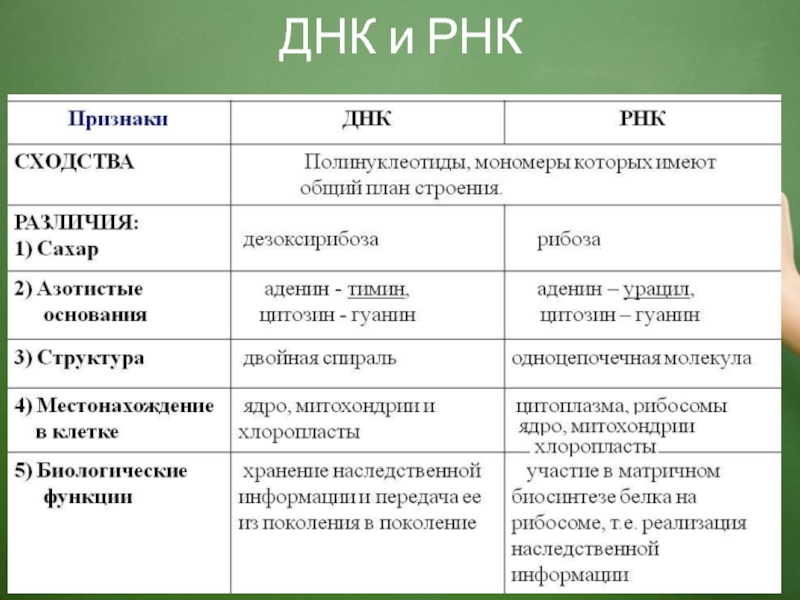 Результаты можно увидеть в виде аллювиальных участков в дополнительном файле 2: рисунки S4, S12 и S13, демонстрирующие, что клонирование очень устойчиво к отбору входных генов и что в целом до 60% наименее вариабельных генов могут быть удалены до задания клонов начинают существенно меняться.
Результаты можно увидеть в виде аллювиальных участков в дополнительном файле 2: рисунки S4, S12 и S13, демонстрирующие, что клонирование очень устойчиво к отбору входных генов и что в целом до 60% наименее вариабельных генов могут быть удалены до задания клонов начинают существенно меняться.
Далее мы оценили устойчивость назначений клонов clonealign к случайному удалению генов для наборов данных SA501, OV2295 и TOV2295. Для каждого мы удалили долю (0,3, 0,5, 0,7, 0,9) генов случайным образом в 10 повторах и вычислили точность и полноту, как если бы совпадения с использованием всех генов представляли истинные клональные назначения. Хотя результаты показывают снижение согласованности с увеличением числа удаленных генов и изменчивостью между наборами данных, в целом до 30% генов можно было бы удалить, чтобы сохранить среднюю точность и полноту> 0.8 для всех клонов (дополнительный файл 2: рисунки S4, S15, S16).
Чтобы ранжировать гены по доле дисперсии, объясняемой клональностью в SA501, полный набор данных был разделен на подмножества, чтобы удалить все рибосомные гены и гены на Х-хромосоме (из-за потери всей хромосомы). Кроме того, мы рассмотрели только гены, дисперсия логарифмической экспрессии которых была больше, чем средняя дисперсия по всем генам, чтобы избежать ложных ассоциаций (т. Е. Если ген экспрессируется только в одной клетке, вся вариация его экспрессии тривиально объясняется клональностью).Доля изменчивости экспрессии была рассчитана с использованием функции aov в R. Затем был проведен анализ обогащения набора генов с использованием пакета fgsea [43] с использованием всех путей ReactomeDB с генами, ранжированными в соответствии с долей изменчивости экспрессии, объясняемой клональностью.
Кроме того, мы рассмотрели только гены, дисперсия логарифмической экспрессии которых была больше, чем средняя дисперсия по всем генам, чтобы избежать ложных ассоциаций (т. Е. Если ген экспрессируется только в одной клетке, вся вариация его экспрессии тривиально объясняется клональностью).Доля изменчивости экспрессии была рассчитана с использованием функции aov в R. Затем был проведен анализ обогащения набора генов с использованием пакета fgsea [43] с использованием всех путей ReactomeDB с генами, ранжированными в соответствии с долей изменчивости экспрессии, объясняемой клональностью.
Клеточные линии и препараты тканей
Клетки OV2295 и TOV2295 культивировали в среде Игла, модифицированной Дульбекко, с добавлением 10% FBS. Ксенотрансплантаты пациентов были созданы в соответствии с протоколом репозитория опухолевой ткани (TTR-H06-00289), который соответствует требованиям Совета по этике исследований UBC BCCA.Все исследования на животных были одобрены Комитетом по уходу за животными Университета Британской Колумбии. Ксенотрансплантаты трансплантировали подкожно самкам мышей NOD/SCID с нулевым гамма-рецептором интерлейкина-2 (NSG) и NOD Rag-1 с нулевым гамма-рецептором интерлейкина-2 (NRG), как описано ранее (Eirew et al., 2015). Собранные опухоли жизнеспособно замораживали в среде DMEM, содержащей 45% FBS и 6% DMSO.
Ксенотрансплантаты трансплантировали подкожно самкам мышей NOD/SCID с нулевым гамма-рецептором интерлейкина-2 (NSG) и NOD Rag-1 с нулевым гамма-рецептором интерлейкина-2 (NRG), как описано ранее (Eirew et al., 2015). Собранные опухоли жизнеспособно замораживали в среде DMEM, содержащей 45% FBS и 6% DMSO.
Секвенирование РНК одиночных клеток Размороженные образцы расщепляли в течение 2 часов коллагеназой/гиалуронидазой, а отдельные клетки подвергали FACS-сортировке на жизнеспособность по отрицательности йодистого пропидия.Суспензии отдельных клеток загружали в контроллер для отдельных клеток 10X genomics, а библиотеки готовили в соответствии со стандартным протоколом набора Chromium Single Cell 3” Reagent v2 Chemistry. Затем библиотеки секвенировали на Illumina Nextseq500/550 с парными считываниями концов по 42 п.н. Cell Ranger 2.0 использовался для выполнения демультиплексирования, выравнивания и подсчета.
Секвенирование ДНК одиночных клеток Суспензии одиночных клеток окрашивали красителями LIVE/DEAD Fixable Red Dead Cell Stains (ThermoFisher) и с использованием красителя cellenONE (Cellenion), отдельные клетки распределяли в каждую лунку нанолуночного чипа, содержащего два уникальных двойных индекса [11]. Библиотеки были созданы с использованием химии транспозазы в одном сосуде (набор для подготовки ДНК-библиотеки Nextera, Illumina), как описано ранее [1, 11]. Вкратце, клетки с пятнами лизировали в течение ночи с последующим мечением, нейтрализацией и ПЦР с индексом образца.
Библиотеки были созданы с использованием химии транспозазы в одном сосуде (набор для подготовки ДНК-библиотеки Nextera, Illumina), как описано ранее [1, 11]. Вкратце, клетки с пятнами лизировали в течение ночи с последующим мечением, нейтрализацией и ПЦР с индексом образца.
Одноклеточная и маловходная РНК-Seq | Преимущества секвенирования отдельных клеток
Введение в секвенирование одноклеточной РНК
Сложные биологические системы определяются согласованными функциями отдельных клеток.Обычные методы, предоставляющие объемные данные генома или транскриптома, не могут выявить клеточная неоднородность, которая движет этой сложностью. Одноклеточное секвенирование — это новое поколение метод секвенирования (NGS), который исследует геномы или транскриптомы отдельных клеток, обеспечивая просмотр вариаций от клетки к клетке в высоком разрешении.
Высокочувствительные методы секвенирования РНК со сверхнизкими затратами и одноклеточной РНК (RNA-Seq) позволяют исследователям
изучить различную биологию отдельных клеток в сложных тканях и понять клеточную
реакции субпопуляции на сигналы окружающей среды.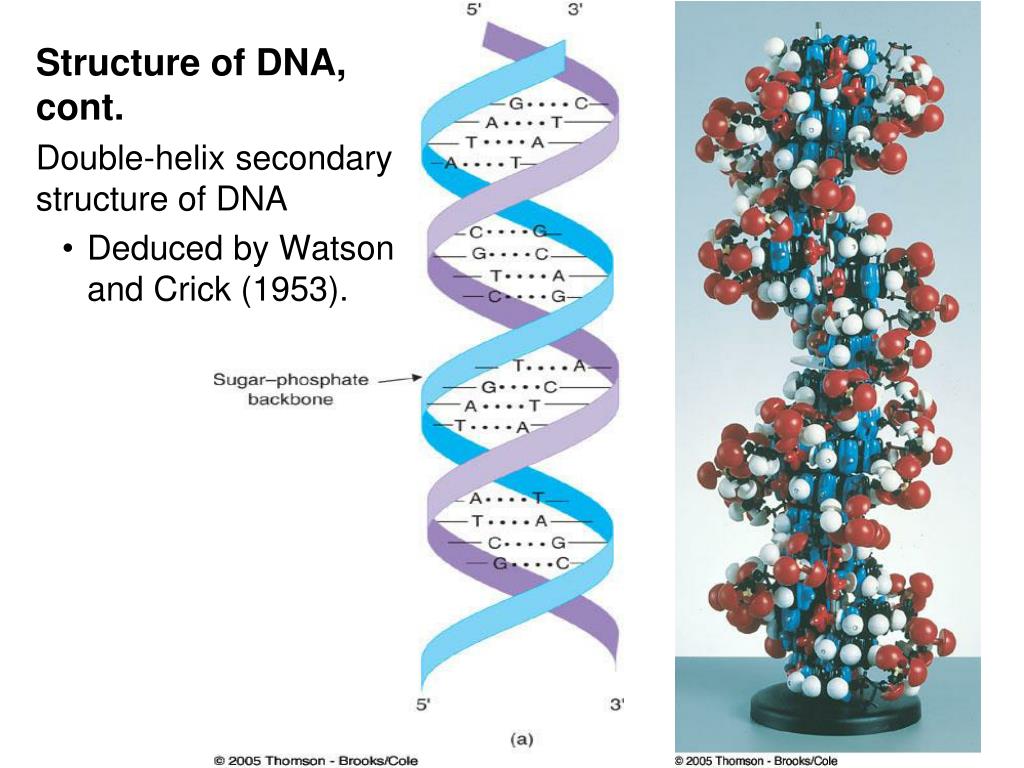 Эти анализы улучшают изучение функции клеток и
гетерогенность в зависимых от времени процессах, таких как дифференцировка, пролиферация и онкогенез.
Эти анализы улучшают изучение функции клеток и
гетерогенность в зависимых от времени процессах, таких как дифференцировка, пролиферация и онкогенез.
Методы секвенирования отдельных клеток
Контроль качества для экспериментов по секвенированию отдельных клеток
Узнайте о лучших методах приготовления клеточных суспензий с помощью растворов для подготовки образцов от Miltenyi Biotec.
Посмотреть вебинарРасширение возможностей экспериментов с отдельными ячейками с мультиплексированием
Исследователи из Калифорнийского университета в Сан-Франциско обсуждают MULTI-Seq, стратегию штрих-кодирования образцов для секвенирования одноклеточной и одноядерной РНК.
Посмотреть вебинарКомбинаторные одноклеточные технологии
Мы выделяем несколько приложений полностью поддерживаемых рабочих процессов, которые помогут вам перейти от суспензий отдельных клеток к анализу данных.
Посмотреть вебинар
Исследовательские статьи о секвенировании отдельных клеток
Институт Вейцмана использует NovaSeq для исследования одиночных клеток
Анализируя одну клетку за раз, профессор Амит улучшает наше понимание биологических систем в
здоровье и болезнь.
Исследование микроокружения опухоли
Секвенирование отдельных клеток оказалось неоценимым при обнаружении внутриклеточной связи в опухолях.
Читать интервьюОбзор исследования одноклеточных
См. обзор рецензируемых публикаций, в которых используется технология Illumina для секвенирования отдельных клеток.
Читать обзор
Высокопроизводительный рабочий процесс для сверхмалого ввода и одноклеточной РНК-Seq
Получите ценную информацию об экспрессии генов с помощью этого чувствительного, масштабируемого и экономичного высокопроизводительного рабочего процесса.
10x Genomics Экспрессия гена одноклеточного хрома
Chromium Single Cell Gene Expression обеспечивает экспрессию 3′-гена транскриптома одной клетки и мультиомные возможности для профилирования десятков тысяч клеток. Исследуйте клеточную гетерогенность, новые мишени и биомаркеры с помощью комбинированной экспрессии генов, экспрессии поверхностных белков или изменений CRISPR в каждой клетке.
Исследуйте клеточную гетерогенность, новые мишени и биомаркеры с помощью комбинированной экспрессии генов, экспрессии поверхностных белков или изменений CRISPR в каждой клетке.
Антитела BioLegend
Олигоконъюгированные антителаTotalSeq™ позволяют измерять белки на уровне отдельных клеток и легко интегрируются в существующие рабочие процессы секвенирования РНК в отдельных клетках, обеспечивая высокомультиплексные исследования белков в отдельных клетках для новых приложений в прецизионной медицине, онкологии, иммунологии, неврологии, и исследования стволовых клеток.
Система NextSeq 550
Гибкая мощность. Скорость и простота для повседневной геномики.
Системы NextSeq 1000 и 2000
75 прорывных инноваций и наш самый простой рабочий процесс. Экономичная гибкость для новых и появляющихся приложений.
Система NovaSeq 6000
Масштабируемая производительность и гибкость практически для любого генома, метода секвенирования и масштаба проекта.
Средство сравнения платформ
Сравните платформы для секвенирования и выберите лучшую систему для своей лаборатории и задач.
Приложение BaseSpace SureCell RNA Single Cell
Простое, но мощное приложение позволяет упростить первичный и вторичный анализ данных одноклеточной РНК-Seq.
Конвейер одноклеточной РНК DRAGEN
Этот интегрированный конвейер идет от базовых вызовов к матрице экспрессии клеток за генами с единой точкой взаимодействия пользователя, совместимой с различными наборами для подготовки образцов сторонних производителей. Доступно в системе NextSeq 2000, а также на сервере DRAGEN.
Рабочий процесс с низкой пропускной способностью для сверхнизкой производительности и одноклеточной РНК-Seq
Описанный ниже низкопроизводительный метод рекомендуется для исследователей, которые хотят обработать небольшое количество клеток для конкретного исследования, например, от десятков до нескольких сотен клеток за эксперимент.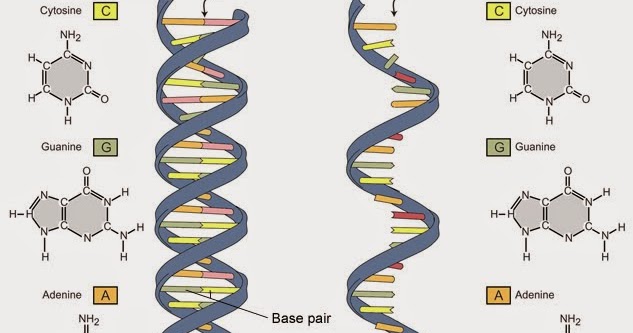
Набор SMART-Seq® Ultra® с низким уровнем ввода РНК
Синтез полноразмерной кДНК из 1–1000 целых клеток или 10 пг–10 нг высококачественной тотальной РНК. Разработан Clontech, продается Takara и оптимизирован для секвенирования Illumina.
Система NextSeq 550
Гибкая мощность. Скорость и простота для повседневной геномики.
Системы NextSeq 1000 и 2000
75 прорывных инноваций и наш самый простой рабочий процесс. Экономичная гибкость для новых и появляющихся приложений.
Система NovaSeq 6000
Масштабируемая производительность и гибкость практически для любого генома, метода секвенирования и масштаба проекта.
Средство сравнения платформ
Сравните платформы для секвенирования и выберите лучшую систему для своей лаборатории и задач.
Руководство по iPathway
Дифференциальная экспрессия генов, взаимодействие лекарств и анализ заболеваний.
Концентратор секвенирования BaseSpace
Генетическая вычислительная среда Illumina для анализа и управления данными NGS.
Механизм корреляции BaseSpace
Растущая библиотека тщательно отобранных геномных данных для помощи исследователям в определении механизмов заболеваний, мишеней для лекарств и биомаркеров.
Конвейер одноклеточной РНК DRAGEN
Этот интегрированный конвейер идет от базовых вызовов к матрице экспрессии клеток за генами с единой точкой взаимодействия пользователя, совместимой с различными наборами для подготовки образцов сторонних производителей. Доступно в системе NextSeq 2000, а также на сервере DRAGEN.
* Расчеты данных в файле.Illumina, Inc., 2015
** Расчеты данных в файле. Иллюмина, Инк., 2020
Глубокое погружение в реальность клеточной РНК
Новый высокочувствительный, специфичный и мультиплексируемый метод обнаружения РНК совершенствует
in situ транскриптомику с потенциалом для ряда биомедицинских примененийBy Benjamin Boettner
Исследователи сравнили метод BOLORAMIS с анализом одиночных молекул FISH (smFISH), общим стандартом для исследований локализации РНК, отследив расположение длинной некодирующей молекулы РНК, известной как MALAT1. Принимая во внимание, что анализ smFISH выявляет MALAT1 довольно размыто, BOLORAMIS разрешает отдельные сайты MALAT1 в клетках (оранжевые на левых изображениях) как маленькие точки и показывает, что перемещение РНК между цитоплазмой (вверху) и ядром (внизу) в клетках . Ядра окрашены в синий цвет на всех флуоресцентных изображениях слева, а изображения справа показывают, где в общих структурах клеток находится MALAT1. Предоставлено: Институт Висса при Гарвардском университете
Принимая во внимание, что анализ smFISH выявляет MALAT1 довольно размыто, BOLORAMIS разрешает отдельные сайты MALAT1 в клетках (оранжевые на левых изображениях) как маленькие точки и показывает, что перемещение РНК между цитоплазмой (вверху) и ядром (внизу) в клетках . Ядра окрашены в синий цвет на всех флуоресцентных изображениях слева, а изображения справа показывают, где в общих структурах клеток находится MALAT1. Предоставлено: Институт Висса при Гарвардском университете (БОСТОН) — Человеческие клетки обычно транскрибируют половину своих примерно 20 000 генов в молекулы РНК в любой момент времени.Как и в случае с белками, функция этих видов РНК зависит не только от их количества, но и от их точной локализации в трехмерном пространстве каждой клетки. Многие молекулы РНК передают информацию о генах из ядра клетки к механизму синтеза белка, распределенному по всей цитоплазме (информационная РНК или мРНК), другие являются компонентами самого этого механизма, в то время как другие молекулы регулируют гены и их экспрессию или выполняют функции, которые остаются неизменными. быть обнаруженным. Важно отметить, что многие заболевания, включая рак и неврологические заболевания, имеют сигнатуры, которые проявляются в виде изменений количества и распределения РНК.
быть обнаруженным. Важно отметить, что многие заболевания, включая рак и неврологические заболевания, имеют сигнатуры, которые проявляются в виде изменений количества и распределения РНК.
Чтобы обеспечить анализ полного набора клеточных РНК, известного как их транскриптом, в их трехмерном пространстве (пространственная транскриптомика), синтетические биологи Института Висса под руководством члена основного факультета Джорджа Черча, доктора философии. в 2014 году сообщили о FISSEQ, эффективной технологии пространственного секвенирования, которая способна одновременно считывать последовательности тысяч этих РНК и визуализировать их трехмерные координаты. Однако мощная способность FISSEQ секвенировать такое большое количество РНК-мишеней на месте имеет свою цену: его эффективность обнаружения и чувствительность для многих из них относительно низки, особенно когда их экспрессия низкая в начале или снижена при заболевании.
Теперь команда церкви разработала новый метод обнаружения РНК по имени Болорамис (короткие для » B Arcoded O LigoNucleotides L Iged O N R Na A Mplified для M Ultiplexed и Parallel I n S ITU анализы»), который решает эту проблему. BOLORAMIS позволяет спроектировать и использовать новый тип ДНК-зонда, который напрямую связывается со своей РНК-мишенью и позволяет прямо синтезировать ампликон ДНК со штрих-кодом, который можно визуализировать с помощью флуоресцентной in situ гибридизации (FISH) или секвенирования in situ. .BOLORAMIS позволяет анализировать различные классы РНК с более высокой специфичностью и чувствительностью, чем FISSEQ и другие методы, работает в контексте клеток и тканей и может быть мультиплексирован. Исследование опубликовано в журнале Nucleic Acids Research .
BOLORAMIS позволяет спроектировать и использовать новый тип ДНК-зонда, который напрямую связывается со своей РНК-мишенью и позволяет прямо синтезировать ампликон ДНК со штрих-кодом, который можно визуализировать с помощью флуоресцентной in situ гибридизации (FISH) или секвенирования in situ. .BOLORAMIS позволяет анализировать различные классы РНК с более высокой специфичностью и чувствительностью, чем FISSEQ и другие методы, работает в контексте клеток и тканей и может быть мультиплексирован. Исследование опубликовано в журнале Nucleic Acids Research .
«С помощью BOLORAMIS мы решили некоторые проблемы, с которыми сталкиваются технологии в области пространственной транскриптомики. Это дает нам значительное преимущество для понимания поведения молекулярных сетей в нормальных и патологических процессах, а также для исследования новых мишеней для лекарств и разработки клинической диагностики в нативном контексте тканей, которые мы теперь можем использовать», — сказал Черч, который является руководитель платформы синтетической биологии Института Висса и профессор генетики в Гарвардской медицинской школе (HMS), а также медицинских наук и технологий в Гарварде и Массачусетском технологическом институте.
«Сила BOLORAMIS заключается в том, что его оптимизированные зонды имеют очень короткий след на РНК, и что он устраняет необходимость сначала генерировать ДНК-реплику молекул РНК на этапе «обратной транскриптазы», который может производить дает неспецифические результаты и стоит дорого», — сказал соавтор Сонглей Лю, аспирант, работающий в команде Черча.
В FISSEQ все РНК сначала фиксируются на месте, а затем вся последовательность РНК копируется в комплементарную последовательность ДНК (обратная транскрипция), которая затем циркулирует и амплифицируется в более крупные шарики ДНК.Их можно секвенировать и визуализировать под специальным флуоресцентным микроскопом. «В BOLORAMIS, минуя этот этап обратной транскрипции и напрямую усиливая сигнал РНК, мы уменьшаем неспецифические и ложные флуоресцентные сигналы», — сказал Лю.
Зонды BOLORAMIS прочно и с высокой специфичностью связываются, как висячий замок, с небольшой последовательностью всего из 25 нуклеотидов в молекуле РНК и, таким образом, имеют гораздо меньший след, чем другие методы направленной пространственной транскриптомики, что улучшает разрешение.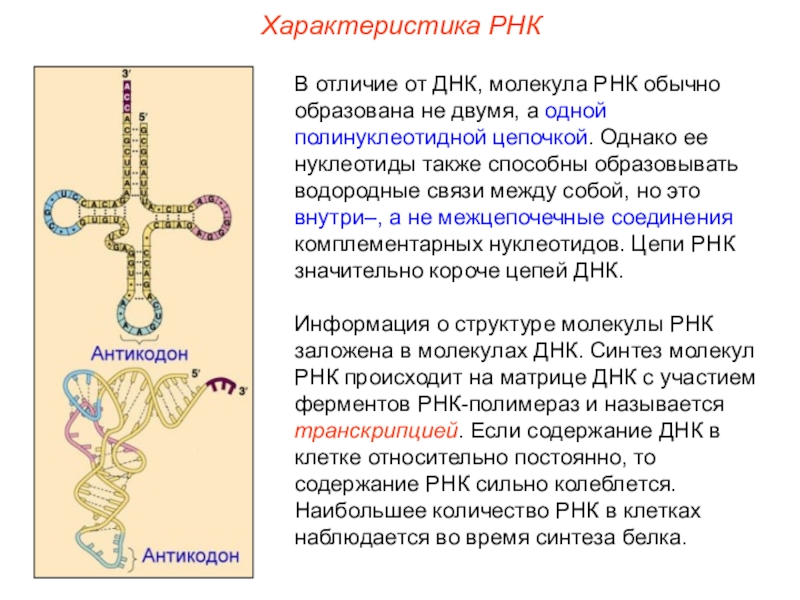 Кроме того, зонды содержат последовательности штрих-кода, которые присваивают уникальный молекулярный почтовый индекс каждому виду РНК-мишени. После гибридизации зондов висячих замков со штрих-кодом с РНК они циркулируют и амплифицируются в крошечный ампликон в месте расположения РНК-мишени без необходимости обратного этапа транскрипции. Затем ампликон можно секвенировать in situ или локализовать с высокой чувствительностью с помощью второго типа зонда, известного как флуоресцентный гибридизационный (FISH) зонд In Situ , который распознает несколько штрих-кодов, содержащихся в одном ампликоне.
Кроме того, зонды содержат последовательности штрих-кода, которые присваивают уникальный молекулярный почтовый индекс каждому виду РНК-мишени. После гибридизации зондов висячих замков со штрих-кодом с РНК они циркулируют и амплифицируются в крошечный ампликон в месте расположения РНК-мишени без необходимости обратного этапа транскрипции. Затем ампликон можно секвенировать in situ или локализовать с высокой чувствительностью с помощью второго типа зонда, известного как флуоресцентный гибридизационный (FISH) зонд In Situ , который распознает несколько штрих-кодов, содержащихся в одном ампликоне.
. Группа впервые исследовала количественные возможности BOLORAMIS в индуцированных человеком плюрипотентных стволовых клетках (ИПСК) путем количественного определения уровней 77 мРНК, кодирующих ряд факторов транскрипции, и 77 некодирующих РНК с другими функциями в генах. регулирование.В то время как BOLORAMIS постоянно демонстрировал высокую и локализованную экспрессию РНК, которая коррелирует со «стволовостью» в клетках, он обнаруживал низкую экспрессию РНК, которые способствуют дифференцировке. В линии раковых клеток BOLORAMIS смог количественно проследить местоположение и движения некодирующей РНК, известной как MALAT1, которая перемещается между двумя различными субклеточными местоположениями, ядром и цитоплазмой. Кроме того, исследователи продемонстрировали, что BOLORAMIS может с высокой чувствительностью отслеживать общую РНК в клетках, расположенных в гораздо более сложной тканевой среде культивируемых органоидов головного мозга человека.
Группа впервые исследовала количественные возможности BOLORAMIS в индуцированных человеком плюрипотентных стволовых клетках (ИПСК) путем количественного определения уровней 77 мРНК, кодирующих ряд факторов транскрипции, и 77 некодирующих РНК с другими функциями в генах. регулирование.В то время как BOLORAMIS постоянно демонстрировал высокую и локализованную экспрессию РНК, которая коррелирует со «стволовостью» в клетках, он обнаруживал низкую экспрессию РНК, которые способствуют дифференцировке. В линии раковых клеток BOLORAMIS смог количественно проследить местоположение и движения некодирующей РНК, известной как MALAT1, которая перемещается между двумя различными субклеточными местоположениями, ядром и цитоплазмой. Кроме того, исследователи продемонстрировали, что BOLORAMIS может с высокой чувствительностью отслеживать общую РНК в клетках, расположенных в гораздо более сложной тканевой среде культивируемых органоидов головного мозга человека.
«Для нас было крайне важно иметь возможность использовать BOLORAMIS для мультиплексного анализа многих РНК, что, как мы надеялись, позволит новая конструкция зонда», — сказал соавтор Суканья Пунтамбакер, доктор философии, научный сотрудник. в команде Черча. Действительно, пакет программного обеспечения, разработанный соавтором Эндрю Павловски, предсказывает идеальные зонды для любой последовательности генов, которые затем могут быть синтезированы в виде полных библиотек для конкретных целей. Команда сделала этот инструмент доступным на сервере GitHub.«Мы использовали систему совместного культивирования нейронных клеток и микроглии мозга, которые, как известно, взаимодействуют во многих нормальных и болезненных процессах, и одновременно нацеливались на 96 различных матричных РНК», — сказал Пунтамбейкер. «Это позволило нам раскрыть пространственные отношения между конкретными клетками и РНК».
в команде Черча. Действительно, пакет программного обеспечения, разработанный соавтором Эндрю Павловски, предсказывает идеальные зонды для любой последовательности генов, которые затем могут быть синтезированы в виде полных библиотек для конкретных целей. Команда сделала этот инструмент доступным на сервере GitHub.«Мы использовали систему совместного культивирования нейронных клеток и микроглии мозга, которые, как известно, взаимодействуют во многих нормальных и болезненных процессах, и одновременно нацеливались на 96 различных матричных РНК», — сказал Пунтамбейкер. «Это позволило нам раскрыть пространственные отношения между конкретными клетками и РНК».
. «В будущих исследованиях высокая функциональность BOLORAMIS в сложных человеческих тканях и специфических для человека органоидах вполне может дать нам преимущество в расшифровке РНК-сигнатур, связанных с неврологическими расстройствами», — добавила старший научный сотрудник Дженни Тэм, доктор философии.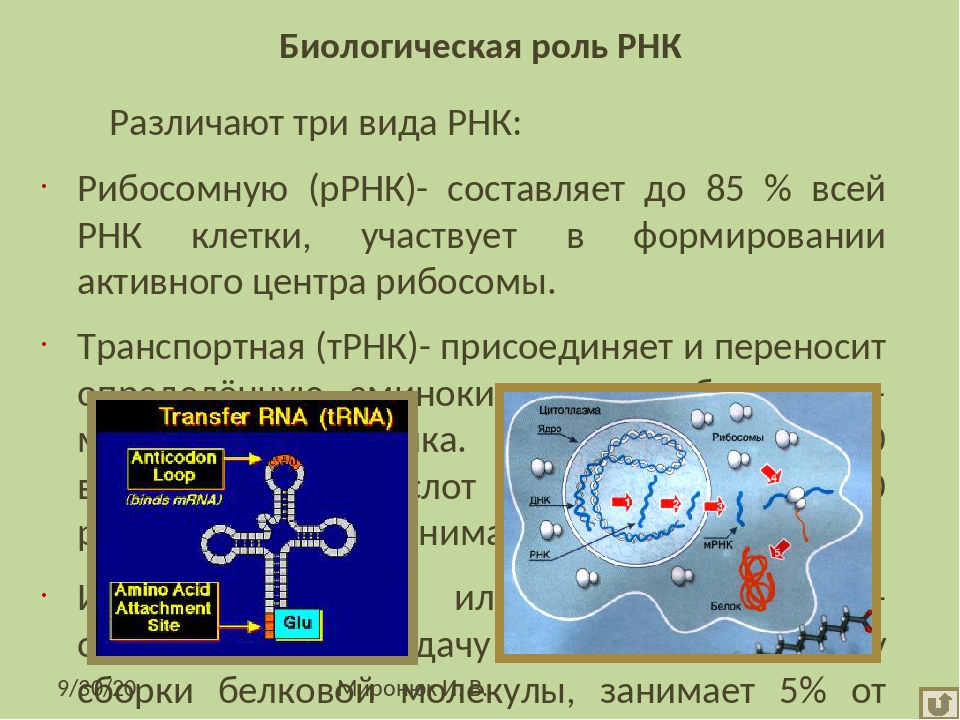 D., который стал соавтором исследования и объединил некоторые исследовательские мероприятия Черча в Институте Висса.
D., который стал соавтором исследования и объединил некоторые исследовательские мероприятия Черча в Институте Висса.
«Оценка точного местоположения и уровней молекул РНК в целых клетках с большей эффективностью и точностью, которую обеспечивает метод BOLORAMIS, должна значительно улучшить нашу способность понять, как организация клеток и тканей влияет на нормальную физиологию, а также на сложные болезненные состояния, и, следовательно, облегчить разработка новых терапевтических и диагностических средств для бесчисленных применений», — сказал директор-основатель Wyss Дональд Ингбер, М.доктор философии, который также является профессором Judah Folkman сосудистой биологии в HMS и Бостонской детской больнице и профессором биоинженерии в Гарвардской школе инженерии и прикладных наук имени Джона А. Полсона.
George ChurchС помощью BOLORAMIS мы решили некоторые проблемы, с которыми сталкиваются технологии в области пространственной транскриптомики. Это дает нам значительное преимущество для понимания поведения молекулярных сетей в нормальных и патологических процессах, а также для исследования новых мишеней для лекарств и разработки клинической диагностики в нативном контексте тканей, которую мы теперь можем использовать.

Другими авторами исследования являются Эсвар Айер, доктор философии, Томас Ферранте, Даниэль Гудвин, Даниэль Фюрт, доктор философии, Кунал Джиндал, Лорен Миффлин, Шахар Алон, доктор философии, Анубхав Синха, Асмамау Васси, доктор философии, Фей Чен, доктор философии, Энн Ченг, Валери Уиллок, Катарина Мейер, доктор философии, Кинг-Хва Линг, доктор философии, Конор Кэмплиссон, Ричи Кохман, доктор философии. , Джон Аах, доктор философии, Дже Хюк Ли, доктор медицины, доктор философии, Брюс Янкнер, доктор философии, профессор генетики и неврологии в HMS, и Эдвард Бойден, доктор философии, профессор кафедры MIT Мозг и когнитивные науки, медиаискусство и наука и биологическая инженерия.Он финансировался Национальным институтом здравоохранения в рамках гранта № RM1 HG008525 и 5R01Mh213279-04.
Новое открытие показывает, что клетки человека могут записывать последовательности РНК в ДНК – бросает вызов центральному принципу биологии
В ходе открытия, бросающего вызов давним догмам в биологии, исследователи показали, что клетки млекопитающих могут преобразовывать последовательности РНК обратно в ДНК, что более характерно для вирусов, чем для эукариотических клеток.
Клетки содержат механизм, который дублирует ДНК в новый набор, который входит во вновь образованную клетку.Тот же класс машин, называемый полимеразами, также создает РНК-сообщения, которые подобны заметкам, скопированным из центрального репозитория ДНК с рецептами, чтобы их можно было более эффективно считывать в белки. Но считалось, что полимеразы работают только в одном направлении ДНК в ДНК или РНК. Это предотвращает переписывание сообщений РНК обратно в основную книгу рецептов геномной ДНК. Теперь исследователи из Университета Томаса Джефферсона предоставили первое доказательство того, что сегменты РНК могут быть записаны обратно в ДНК, что потенциально бросает вызов центральной догме биологии и может иметь широкие последствия, затрагивающие многие области биологии.
«Эта работа открывает двери для многих других исследований, которые помогут нам понять важность наличия механизма преобразования РНК-сообщений в ДНК в наших собственных клетках», — говорит Ричард Померанц, доктор философии, доцент кафедры биохимии и молекулярной биологии Томаса Джефферсона. Университет. «Реальность того, что человеческая полимераза может делать это с высокой эффективностью, вызывает много вопросов». Например, это открытие предполагает, что сообщения РНК могут использоваться в качестве шаблонов для восстановления или перезаписи геномной ДНК.
Университет. «Реальность того, что человеческая полимераза может делать это с высокой эффективностью, вызывает много вопросов». Например, это открытие предполагает, что сообщения РНК могут использоваться в качестве шаблонов для восстановления или перезаписи геномной ДНК.
Работа была опубликована 11 июня 2021 года в журнале Science Advances.
Вместе с первым автором Гурушанкаром Чандрамули и другими сотрудниками команда доктора Померанца начала с исследования одной очень необычной полимеразы, называемой тета-полимеразой. Из 14 ДНК-полимераз в клетках млекопитающих только три выполняют основную часть работы по дублированию всего генома для подготовки к клеточному делению. Остальные 11 в основном участвуют в обнаружении и восстановлении разрывов или ошибок в цепях ДНК.Тета-полимераза восстанавливает ДНК, но очень подвержена ошибкам и делает много ошибок или мутаций. Поэтому исследователи заметили, что некоторые из «плохих» качеств полимеразы тета были общими с другой клеточной машиной, хотя и более распространенной у вирусов — обратной транскриптазой. Подобно Pol theta, обратная транскриптаза ВИЧ действует как ДНК-полимераза, но также может связывать РНК и считывать РНК обратно в цепь ДНК.
Подобно Pol theta, обратная транскриптаза ВИЧ действует как ДНК-полимераза, но также может связывать РНК и считывать РНК обратно в цепь ДНК.
В серии элегантных экспериментов исследователи протестировали полимеразу тета против обратной транскриптазы ВИЧ, которая является одной из наиболее изученных в своем роде.Они показали, что тета-полимераза способна преобразовывать РНК-сообщения в ДНК, что она делала так же, как и обратная транскриптаза ВИЧ, и что на самом деле она выполняла эту работу лучше, чем при дублировании ДНК в ДНК. Тета-полимераза была более эффективной и вносила меньше ошибок при использовании матрицы РНК для записи новых сообщений ДНК, чем при дублировании ДНК в ДНК, что позволяет предположить, что эта функция может быть ее основной целью в клетке.
Группа сотрудничала с лабораторией доктора Сяоцзяна С. Чена в Университете Южной Калифорнии и использовала рентгеновскую кристаллографию для определения структуры и обнаружила, что эта молекула способна изменять форму, чтобы приспособиться к более объемной молекуле РНК — подвиг, уникальный среди полимераз.
«Наши исследования показывают, что основная функция полимеразы тета — действовать как обратная транскриптаза, — говорит доктор Померанц. «В здоровых клетках целью этой молекулы может быть РНК-опосредованная репарация ДНК. В нездоровых клетках, таких как раковые клетки, тета-полимераза сильно экспрессируется и способствует росту раковых клеток и устойчивости к лекарствам. Будет интересно узнать, как активность тета-полимеразы в отношении РНК способствует восстановлению ДНК и пролиферации раковых клеток».
Ссылка: «Polθ обратно транскрибирует РНК и способствует репарации ДНК на шаблоне РНК» Гурушанкар Чандрамули, Джиемин Чжао, Шейн Макдевитт, Тимур Русанов, Трунг Хоанг, Никита Борисонник, Тейлор Треддиник, Фелиция Уэнсдей Лопезколорадо, Татьяна Кент, Лабиба А.Сиддик, Джозеф Мэллон, Джеклин Хун, Зайнаб Шода, Екатерина Кашкина, Алессандра Брамбати, Джереми М. Старк, Сяоцзян С. Чен и Ричард Т. Померанц, 11 июня 2021 г., Science Advances .
DOI: 10.1126/sciadv. abf1771
abf1771
Это исследование было поддержано грантами NIH 1R01GM130889-01 и 1R01GM137124-01, а также R01CA197506 и R01CA240392. Это исследование также было частично поддержано грантом Tower Cancer Research Foundation. Авторы сообщают об отсутствии конфликта интересов.
«Пусть клетки расскажут историю»
В поисках неизвестного неизвестного
Когда Полсон и Шапюи искали ключевое изменение в раковых клетках ее пациента, они не знали, что они ищут и в какой клеточной популяции они это найдут.Одно из больших преимуществ секвенирования одноклеточной РНК — и то, что отличает его от многих других экспериментальных инструментов — это тот факт, что исследователи могут использовать его для обнаружения различий между клетками или даже совершенно новых клеточных популяций, о существовании которых они не знали раньше. , — сказал Биелас.
Доктор Энтони Ронгво из The Hutch изучает, как рак метастазирует или распространяется по телу, особенно меланома рака кожи. Как и Шапюи и Полсон, его лаборатория воспользовалась преимуществами высокопроизводительного секвенирования РНК одиночных клеток, чтобы найти неизвестную популяцию клеток, которые способствуют метастазированию.
Как и Шапюи и Полсон, его лаборатория воспользовалась преимуществами высокопроизводительного секвенирования РНК одиночных клеток, чтобы найти неизвестную популяцию клеток, которые способствуют метастазированию.
Особое внимание он уделяет макрофагам, типу иммунных клеток. Хотя макрофаги больше всего известны как мусоросборники иммунной системы, собирающие инфицированные клетки, иногда они могут подавлять иммунный ответ. Все больше данных свидетельствует о том, что существует гораздо больше подтипов макрофагов, чем предполагалось изначально, и опухоли часто находят способы превратить макрофаги из борцов с раком в промоутеров рака, которые могут даже блокировать другие иммунные клетки от атаки опухоли.
Эти разные типы макрофагов отличает не их ДНК, а то, что они делают .И эти различия можно найти в их мРНК.
Группа Ронгво объединилась с Биеласом и Готтардо, чтобы использовать высокопроизводительное секвенирование РНК одиночных клеток, чтобы «позволить макрофагам рассказать нам, кто они такие», сказал он.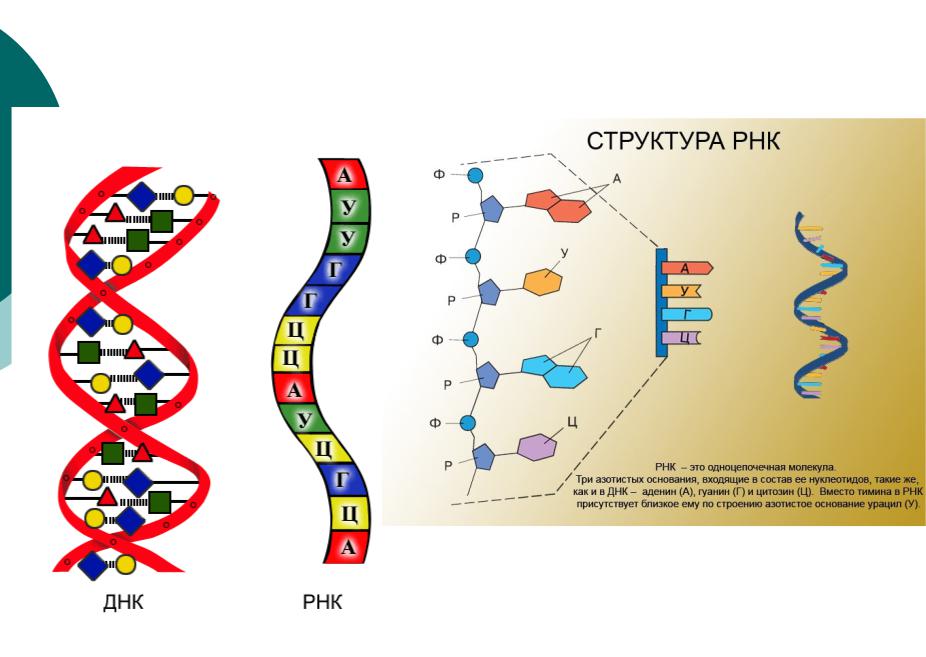
Под руководством Вуайе и доктора Триши Сиппель из лаборатории Ронгво ученые обнаружили ранее не описанную популяцию макрофагов, которые превратились в помощников при опухолях. Полсон, который также внес свой вклад, назвал их «бад-офагами». Эти перебежчики используют особый набор мРНК, что дает исследователям подсказки о том, как плохие фаги выживают в опухолях и как они могут мешать работе иммунных клеток, называемых Т-клетками.
Изучение экологии рака
Доктор Джерри Радич, врач-ученый из Хатча, изучающий лейкемию и работавший с Биеласом над валидацией платформы 10x, изучает лейкемию и ее лечение — кто рецидивирует, а кто излечивается. Но ответы на эти вопросы могут лежать не только в самих раковых клетках.
«Рак — это экосистема со многими [генетическими] клонами, которые могут конкурировать или сотрудничать друг с другом», не говоря уже о других клетках, таких как иммунные клетки, которые взаимодействуют с раковыми клетками, — сказал Радич.Он и его коллега Хатч, доктор Эми Пагириган, были одними из первых, кто исследовал генетику лейкемии на уровне одной клетки, описав гораздо более сложное эволюционное древо рака, чем предполагалось ранее.
С появлением новой технологии секвенирования одноклеточной РНК Радич и множество других исследователей спешат применить ее. Большая способность понимать все компоненты раковой экосистемы — раковые и нераковые — и то, как экосистема меняется после терапии, поможет ученым продвигать и совершенствовать методы лечения.
Возьмем некоторые виды клеточной иммунотерапии, в которых Т-клетки конструируются для нацеливания на опухолевые клетки: у некоторых пациентов болезнь исчезает, но у некоторых болезнь сопротивляется или рецидивирует.
«По экспрессии РНК можно сделать вывод о функции, которая предполагает, что «О, да, в этой популяции людей, которые вылечились, эти Т-клетки действительно активизировались и атакуют опухоль», — сказал Радич. Напротив, возможно, Т-клетки у пациентов, чей рак не реагирует, также имеют отличительные паттерны мРНК: «Вы можете сказать: «Посмотрите на этих ленивых бездельников, они не тянут свою тяжесть.’»
Доктор Марк Хедли из The Hutch изучает, как иммунная система способствует распространению рака. Сосредоточившись конкретно на метастатических опухолях, возникающих в легких, он сосредоточился на самых ранних моментах метастазирования, когда «небольшая группа» иммунных клеток взаимодействует, возможно, только с одной метастатической опухолевой клеткой.
Сосредоточившись конкретно на метастатических опухолях, возникающих в легких, он сосредоточился на самых ранних моментах метастазирования, когда «небольшая группа» иммунных клеток взаимодействует, возможно, только с одной метастатической опухолевой клеткой.
«Моя точка зрения такова, что если вы хотите понять процесс [метастазирования], вам действительно нужно остановиться на уровне одной клетки», — сказал Хедли. Хедли считает, что межклеточная связь между иммунными клетками и метастатическими опухолевыми клетками закладывает основу для будущего, определяя, какие метастатические опухолевые клетки преуспеют в засеве новой опухоли, а также помогая формировать долговременное микроокружение опухоли.
Преобразование идей в клинику
Технологии секвенирования отдельных клеток уже дают информацию, которая может быть использована в клинике. Полсон вместе с Шапюи и членом ее команды доктором Меган Макафи работает над улучшением лечения других больных раком.
Она работает с пациентами с карциномой из клеток Меркеля, или MCC, редким и агрессивным раком кожи.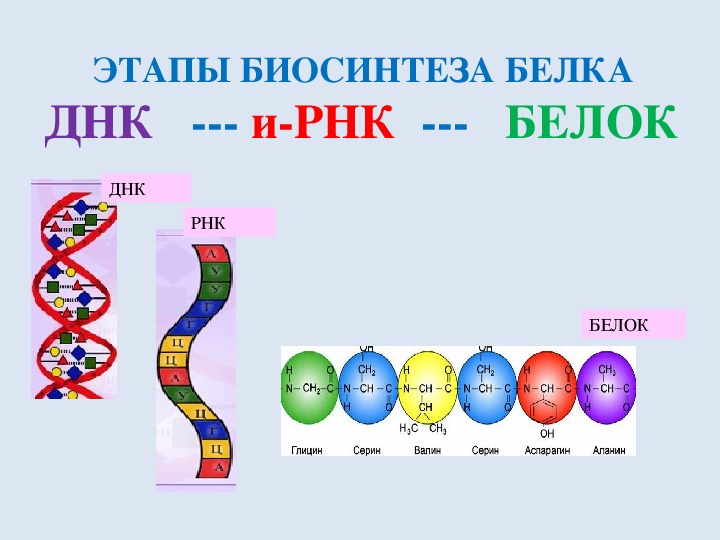 Иммунотерапия изменила уход за этими пациентами, но не у всех опухоль поддается лечению.Клетки, наиболее ответственные за устранение опухолей, — это Т-клетки, которые полагаются на уникальную молекулу, называемую Т-клеточным рецептором, или TCR. Генная инженерия позволила ученым заставить Т-клетки других пациентов экспрессировать TCR из мощной противоопухолевой Т-клетки. С технологией секвенирования РНК одиночных клеток исследователи Фреда Хатча могут найти эти TCR легче, чем когда-либо.
Иммунотерапия изменила уход за этими пациентами, но не у всех опухоль поддается лечению.Клетки, наиболее ответственные за устранение опухолей, — это Т-клетки, которые полагаются на уникальную молекулу, называемую Т-клеточным рецептором, или TCR. Генная инженерия позволила ученым заставить Т-клетки других пациентов экспрессировать TCR из мощной противоопухолевой Т-клетки. С технологией секвенирования РНК одиночных клеток исследователи Фреда Хатча могут найти эти TCR легче, чем когда-либо.
Поле в движении
Поскольку эта область очень молода, ученые все еще пытаются определить, какая платформа лучше всего соответствует их индивидуальным исследовательским потребностям, сказал Хедли.Он использует как платформу 10X, так и сотрудничает с Seelig, чтобы применить свой подход к раздельному пулу. Каждый метод несколько отличается своими требованиями к тому, как обрабатываются ячейки, и к данным, которые он производит. Хедли изучает контексты, в которых каждый подход работает лучше всего.
Методы секвенирования одноклеточной РНК с умеренной пропускной способностью были впервые опубликованы в 2009 г., но в 2015 г. эта область значительно расширилась, когда новые методы, называемые Cyto-seq, Drop-seq и InDrop, позволили секвенировать РНК из десятков или сотни тысяч клеток в одном эксперименте.
Огромный размер наборов данных, созданных с помощью этих методов, стимулирует ряд достижений в науке о данных, поскольку вычислительные биологи разрабатывают более совершенные способы анализа, визуализации, хранения и доступа к этим данным.
Попытка воспользоваться преимуществами новых технологий без учета того, как данные будут генерироваться и анализироваться, может привести к напрасным экспериментам, сказал Готтардо, который возглавляет Интегрированный исследовательский центр трансляционных данных Фреда Хатча и является обладателем премии J.Почетный председатель Фонда Орина Эдсона.
Тесное сотрудничество с вычислительными биологами гарантирует, что исследователи планируют эксперименты так, чтобы можно было ответить на четкие биологические вопросы, сказал он.
В будущем Радич ожидает, что технологии позволят комбинировать секвенирование ДНК и секвенирование РНК из одной и той же клетки, что позволит таким ученым, как он, напрямую связывать функциональную информацию, основанную на РНК, с уникальной генетикой клетки.
В то время как на горизонте ждут новые приложения, Полсон использует возможности, которые она видит в технологиях секвенирования РНК одиночных клеток для продвижения иммунотерапии рака.
«Я думаю, что они действительно произвели революцию в этой области. Они будут полезны для множества различных приложений», — сказал Полсон.
Часто задаваемые вопросы о секвенировании одноклеточной РНК
Общие вопросы
Что такое одноклеточная РНК-Seq?
Single-Cell RNA-Seq обеспечивает профилирование транскрипции тысяч отдельных клеток. Этот уровень анализа пропускной способности позволяет исследователям понять на уровне одной клетки, какие гены экспрессируются, в каких количествах и как они различаются между тысячами клеток в гетерогенной выборке.
Рабочий процесс для Single-Cell RNA-Seq описан ниже. Изолированные клетки замораживаются заказчиком в соответствии с протоколом криоконсервации GENEWIZ и отправляются на ночь. В GENEWIZ оценивается жизнеспособность клеток и, при необходимости, может быть выполнено удаление мертвых клеток (дополнительную информацию см. ниже). Затем клетки обрабатываются контроллером 10x Genomics Chromium™ для создания библиотек отдельных клеток. Затем библиотеки объединяются и секвенируются на платформе Illumina ® .
Внутри контроллера Chromium клетки загружаются на микрожидкостный чип и инкапсулируются в капли, содержащие гелевые шарики со штрих-кодом и реагенты для обратной транскрипции.После лизиса клеток шарики захватывают поли(А)-хвосты молекул мРНК. Обратная транскрипция генерирует кДНК, помеченную штрих-кодом 10x для идентификации клетки и уникальным молекулярным идентификатором (UMI) для маркировки транскрипта мРНК. Объединенная кДНК амплифицируется в большом количестве, и к фрагментам добавляются адаптеры Illumina, включая образец индекса, для создания библиотек секвенирования.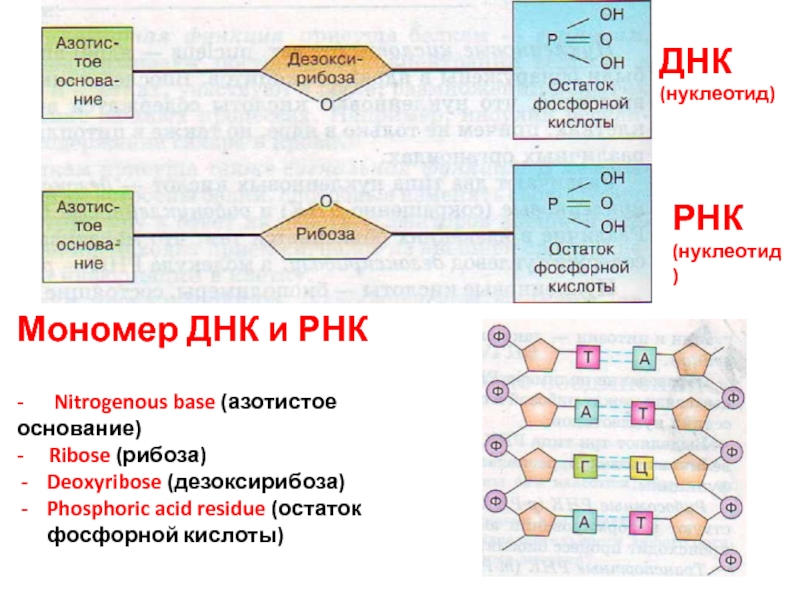
Прочтите нашу статью «Как работает секвенирование отдельных клеток» для получения дополнительной информации.
2. Чем Single Cell RNA-Seq отличается от Standard и Ultra-Low Input RNA-Seq?
Standard RNA-Seq дает репрезентативный снимок состояния транскрипции, усредненный по всем клеткам.Недостатком традиционной РНК-Seq является разрешение отдельных клеток и потеря клеточных субпопуляций. Single-Cell RNA-Seq позволяет исследователям не только идентифицировать клеточные субпопуляции, но и полностью исследовать их на уровне отдельных клеток в гетерогенном образце.
Подобно стандартному RNA-Seq, RNA-Seq со сверхнизким входом обеспечивает массовый анализ экспрессии всей клеточной популяции; однако, как следует из названия, используется очень ограниченное количество исходного материала, всего 10 пг или несколько клеток.Для одноклеточной РНК-Seq требуется не менее 50 000 клеток (рекомендуется 1 миллион) в качестве исходных данных. Подробнее о правилах подачи образцов см. ниже.
ниже.
Прочтите нашу статью «3 основных фактора, которые следует учитывать перед началом проекта секвенирования отдельных клеток», чтобы узнать, является ли секвенирование одиночных клеток подходящим подходом NGS для вашего эксперимента.
3. Каков наш рабочий процесс обработки образцов?
Здесь показан наш рабочий процесс. Жизнеспособность и количество клеток измеряют сразу после получения образца.Чтобы обеспечить высочайшее качество данных, клетки должны быть обработаны в системе 10x Genomics Chromium как можно скорее. Таким образом, мы разработали предопределенный рабочий процесс, когда образцы не проходят первоначальный контроль качества на жизнеспособность или количество клеток.
4. Как мертвые клетки влияют на качество данных?
Низкая жизнеспособность клеток вызывает ненадежное восстановление клеток в системе Chromium и обычно приводит к пропущенным целям секвенирования и неоптимальным результатам. Кроме того, окружающая РНК, высвобождаемая мертвыми или апоптозными клетками, увеличивает фоновый шум, снижая качество данных. Чтобы максимизировать объем полезных данных секвенирования в проектах с одной клеткой, необходимо свести к минимуму долю мертвых клеток. Мы рекомендуем, чтобы жизнеспособность клеток превышала 90%, но не менее 70%. Образцы с жизнеспособностью менее 70% могут подвергаться удалению мертвых клеток (DCR). Для DCR не требуется минимум 10 6 клеток и никакой предварительной обработки магнитными шариками. Смотрите наш рабочий процесс для получения дополнительной информации.
Чтобы максимизировать объем полезных данных секвенирования в проектах с одной клеткой, необходимо свести к минимуму долю мертвых клеток. Мы рекомендуем, чтобы жизнеспособность клеток превышала 90%, но не менее 70%. Образцы с жизнеспособностью менее 70% могут подвергаться удалению мертвых клеток (DCR). Для DCR не требуется минимум 10 6 клеток и никакой предварительной обработки магнитными шариками. Смотрите наш рабочий процесс для получения дополнительной информации.
5. Как происходит удаление мертвых клеток?
Удаление мертвых клеток (DCR) использует магнитные шарики, конъюгированные с антителами против аннексина V, маркера мертвых и апоптотических клеток (см. ниже).Шарики захватывают мертвые или умирающие клетки, обогащая образец живыми клетками.
6. Сколько клеток можно секвенировать на образец на Chromium Controller?
Для выявления полного разнообразия экспрессии генов в ткани или клеточной популяции часто требуется анализ от сотен до тысяч жизнеспособных клеток. Благодаря нашим рабочим процессам с отдельными клетками исследователи могут указать количество секвенируемых клеток от 500 до 10 000 клеток на образец. Обычно мы рекомендуем ориентироваться на 3000 клеток на образец для большинства экспериментов.
Благодаря нашим рабочим процессам с отдельными клетками исследователи могут указать количество секвенируемых клеток от 500 до 10 000 клеток на образец. Обычно мы рекомендуем ориентироваться на 3000 клеток на образец для большинства экспериментов.
7. Сколько ридов мне нужно для моего эксперимента?
Необходимое количество прочтений зависит от размера генома, количества известных генов, типа клеток и транскриптов. Как правило, мы рекомендуем 100 000 прочтений на ячейку, чтобы максимизировать идентификацию транскриптов.
8. Какой тип анализа данных доступен?
Мы используем программное обеспечение Cell Ranger от 10x Genomics для анализа данных, которое включает следующее:
- Чтение выравнивания
- Матрица характеристик штрих-кода
- Цифровая матрица экспрессии генов
- Кластеризация
- Анализ экспрессии генов
- Проекции t-SNE (показаны ниже)
Образец отчета доступен здесь (требуется авторизация).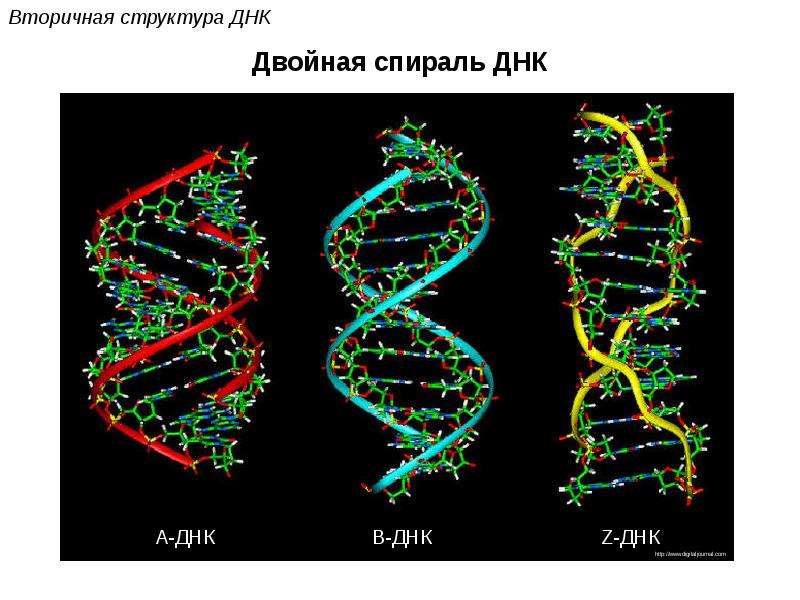
Вы также можете использовать браузер ячеек Loupe от 10x Genomics для изучения своих данных, включая интерактивные инструменты для поиска важных генов, определения типов клеток и изучения субструктуры. Дополнительную информацию, учебные пособия и демонстрации можно найти здесь.
9. Как доставляются данные?
GENEWIZ порекомендует лучший вариант доставки данных на основе платформы и деталей проекта. Варианты доставки данных включают:
- Протокол безопасной передачи файлов (SFTP)
- Облачная учетная запись клиента
- Жесткий диск (может взиматься дополнительная плата)
10.Как будут доставлены мои данные?
- Если ваши результаты отправляются вам на жестком диске, вы можете отследить их доставку, используя номер для отслеживания, указанный в электронном письме о доставке, отправленном от GENEWIZ.
- Если ваши результаты доставляются с помощью другого метода доставки данных, такого как AWS S3, Aspera или других облачных методов , обратитесь к электронному письму о доставке, отправленному от GENEWIZ, для получения подробной информации о передаче и проконсультируйтесь с вашим ИТ-отделом.
 отдела для обеспечения соблюдения политики вашего учреждения.
отдела для обеспечения соблюдения политики вашего учреждения. - Если ваши результаты отправляются через безопасный протокол передачи файлов (sFTP) , обратитесь к Руководству по загрузке данных GENEWIZ по sFTP за инструкциями по загрузке ваших данных и советами по устранению неполадок.
11. Как мне связаться с GENEWIZ для получения технической консультации по моему проекту?
Команда GENEWIZ NGS состоит из доктора философии. ученые, которые могут помочь вам оптимизировать дизайн вашего проекта и дать консультацию. Смотрите нашу контактную информацию внизу страницы.
12. Какова цена одноклеточной РНК-Seq?
Пожалуйста, отправьте запрос на получение точной информации о ценах, так как стоимость зависит от деталей проекта.
Вернуться к началу
Подготовка проб
13. Как подготовить и отправить образцы?
- Тип образца: Замороженная клеточная суспензия в соответствии с протоколом, предоставленным GENEWIZ, представлена в двух экземплярах.
 Также принимаются фиксированные метанолом клетки. Для некоторых образцов может быть доступна диссоциация тканей (запросите, если интересно).
Также принимаются фиксированные метанолом клетки. Для некоторых образцов может быть доступна диссоциация тканей (запросите, если интересно). - Рекомендуемое количество: >10 6 клеток в 1 мл
- Минимальное количество: 50 000 клеток в 500 мкл (Примечание: для удаления мертвых клеток требуется 10 6 клеток)
- Буфер: Среда для криоконсервации (например, среда для культивирования клеток с добавлением 20% FBS и 10% ДМСО)
- Пробирки/планшеты: Для проектов с менее чем 36 образцами подготовьте образцы в микроцентрифужных пробирках с четкой маркировкой. Для более крупных проектов используйте 96-луночные планшеты для ПЦР.
- Способ доставки: Сухой лед на ночь. Отправляйте образцы прямо на наш объект. Используйте адрес доставки, указанный в квитанции о заказе. Примечание. Компания GENEWIZ совместно с 10x Genomics разработала собственный протокол криоконсервации, облегчающий отправку образцов.

14. Что произойдет, если мои образцы не будут соответствовать вашим требованиям к исходному материалу?
Пожалуйста, свяжитесь с нами (см. информацию внизу страницы).
Вернуться к началу
Заказ и обработка
15.Как мне запросить цитату?
Для краткого руководства посмотрите видео ниже. Нажмите значок Single-Cell Services , чтобы перейти к форме запроса цены.
16. Как подтвердить предложение?
Для краткого руководства посмотрите видео ниже.
17. Как отправить образец формы GENEWIZ (чтобы предоставить подробную информацию о моих образцах)?
Для краткого руководства посмотрите видео ниже. >
>
18. Как я могу следить за ходом моего проекта?
Статус вашего заказа, включая предполагаемую дату доставки, можно просмотреть в любое время через вашу учетную запись GENEWIZ. Посетите страницу сводки заказов вашего проекта, чтобы узнать текущий статус заказа.
19. Как долго вы храните образцы?
Мы храним образцы в течение 1 года после завершения проекта. Пожалуйста, свяжитесь с нами, если вы хотите, чтобы образцы хранились дольше или были отправлены вам обратно.
Вернуться к началу
Есть конкретный вопрос?
Электронная почта | Телефон 1-877-GENEWIZ (436-3949), доб.
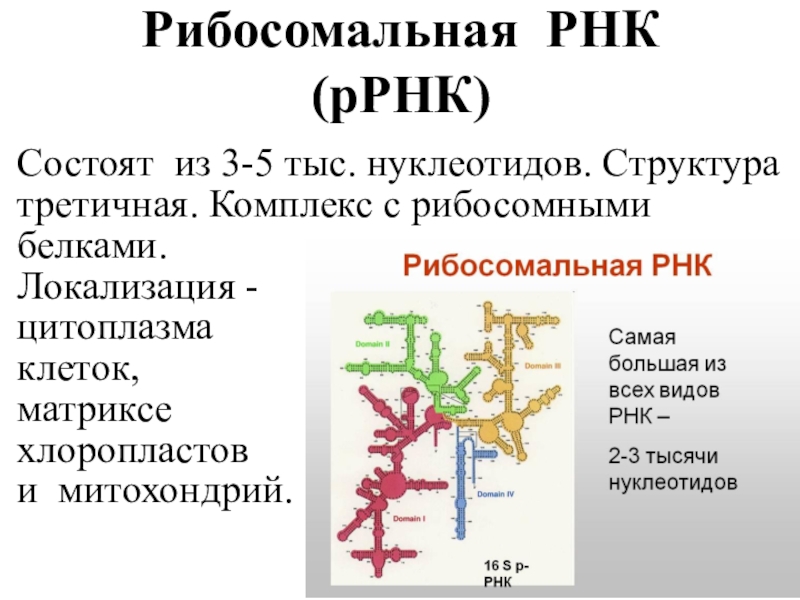 1
1
Вернуться к началу
Научно-исследовательский институт Лернера
О
Миссия Центра геномики Исследовательского института Лернера состоит в том, чтобы обеспечить легкий доступ к самым современным службам ДНК и РНК исследователям Кливлендской клиники, а также внешним академическим и коммерческим пользователям.
В Genomics Core используются передовые технологии геномики для получения высококачественных данных в кратчайшие сроки по доступной цене.Мы предлагаем широкий спектр услуг, включая профилирование экспрессии генов на основе микрочипов Illumina, генотипирование и секвенирование нового поколения (NGS). Наша лаборатория оснащена известным настольным секвенсором Illumina MiSeq и Illumina NovaSeq 6000, которые позволяют проводить быстрый и эффективный целенаправленный анализ всего генома. Мы подготавливаем и нормализуем библиотеки для РНК-секвенирования, микроРНК-секвенирования, ДНК-секвенирования, секвенирования целевого экзома, секвенирования ампликонов, бактериальных 16S-исследований, секвенирования всего генома и секвенирования РНК отдельных клеток. Другие услуги включают контроль качества ДНК/РНК на биоанализаторе Agilent Technologies и системах Tapestation, разделение ДНК и хроматина с помощью технологии Covaris S2 и поддержку анализа данных микрочипов. Мы также сотрудничаем с биорепозиторием геномной медицины для извлечения ДНК и РНК для наших пользователей.
Другие услуги включают контроль качества ДНК/РНК на биоанализаторе Agilent Technologies и системах Tapestation, разделение ДНК и хроматина с помощью технологии Covaris S2 и поддержку анализа данных микрочипов. Мы также сотрудничаем с биорепозиторием геномной медицины для извлечения ДНК и РНК для наших пользователей.
Команда Genomics Core может работать с отдельными пользователями для предоставления услуг, не перечисленных здесь. Мы также предоставляем письма поддержки для заявок на федеральные и нефедеральные гранты и открыты для сотрудничества в исследованиях.Пожалуйста, свяжитесь с Genomics Core по телефону [email protected] или 216.444.7124 для запросов на обслуживание и ежедневного общения. Все научные вопросы или вопросы по дизайну следует направлять доктору Ченгу по телефону [email protected] . Все запросы на письма поддержки заявок на гранты следует направлять доктору Энгу по телефону [email protected] .
Все остальные вопросы можно направлять по адресу LRIgenomics@ccf. org .
org .
Контакты
|
Чарис Энг, доктор медицинских наук |
|
|
Ю-Вей Ченг, доктор философии |
|
|
Dean Horton, PhD |
|
|
Мин Хуэй Лим |
|
|
Эван Уэлч |
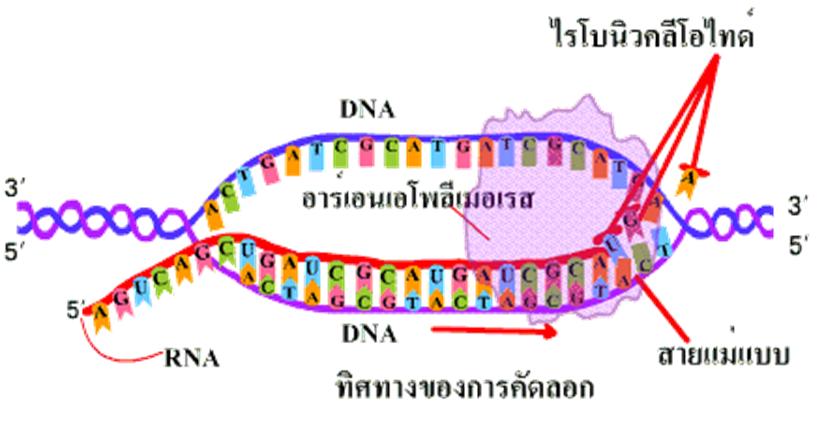 org
org Услуги
Секвенирование нового поколения (NGS)Появление секвенирования нового поколения (NGS) произвело революцию в научных открытиях в различных областях, от исследований болезней человека до наук об окружающей среде и сельском хозяйстве.В расчете на образец NGS значительно дешевле, быстрее, требует значительно меньшего количества ДНК, чем секвенирование по Сэнгеру, и обладает рядом других преимуществ:
- Полное профилирование транскриптома модельных и немодельных организмов с высокой чувствительностью для обнаружения новых транскриптов и вариантов сплайсинга, а также возможность получения специфичных для нитей паттернов экспрессии
- Точное обнаружение SNP при низком покрытии и характеристика структурных перестроек, включая сбалансированные транслокации
- Точная глобальная оценка взаимодействий связывания ДНК и белка
- Эффективное и недорогое микробиомное и метагеномное секвенирование
Услуги NGS, предлагаемые в рамках Genomics Core, включают подготовку библиотек и секвенирование для широкого спектра применений, включая РНК-секвенирование, микроРНК-секвенирование, ДНК-секвенирование, секвенирование отдельных клеток, секвенирование экзома и всего генома, секвенирование ампликонов и бактериальное 16S метагеномика.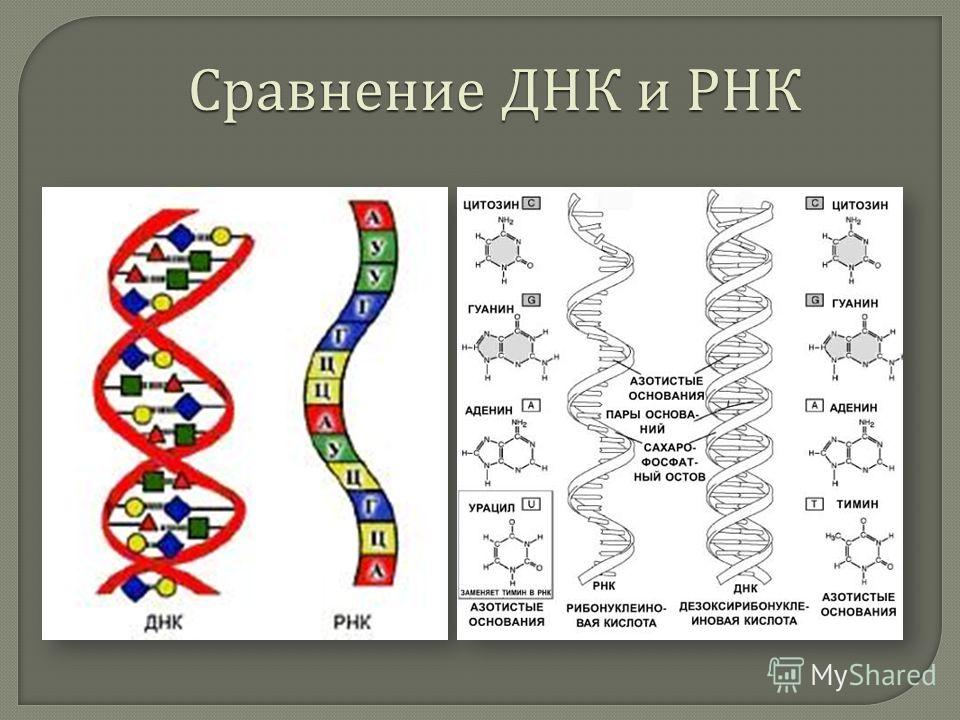
Система Genomics Core оснащена последним обновлением Illumina NovaSeq 6000, а также настольным секвенсором Illumina MiSeq, что позволяет нам предоставлять весь спектр услуг секвенирования следующего поколения по доступной цене и в короткие сроки. NovaSeq 6000 и MiSeq полностью интегрированы с BaseSpace, предлагая автоматизированные рабочие процессы биоинформатики и обмен данными в облаке.
Генотипирование на основе массивов
Пользовательские протоколы гибридизации микрочипов в зависимости от концентрации РНК (от 500 нг/мкл до 500 пг/мкл).РНК
сначала преобразуется в кРНК и гибридизуется с зондами на чипе.
Микросхема Illumina Beadchip
Illumina iScan
- Чипы микрочипов сканируются с помощью Illumina iScan.
- Пользователи предоставляют массивы (человеческие или мышиные) и общую РНК. Core выполнит этап контроля качества РНК, обработает образцы РНК, если они пройдут, гибридизируют с массивами и создадут соответствующие выходные файлы (в Cleveland Clinic команда Core может установить программное обеспечение Illumina GenomeStudio на ПК в вашей лаборатории).

- Предоставляем бесплатные консультации по планированию эксперимента и подготовке образцов.
Библиотека ДНК, РНК и NGS QC
Качество образцов является одним из важнейших факторов, определяющих успех обработки образцов. В Genomics Core используются хорошо зарекомендовавшие себя процедуры контроля качества, чтобы предоставлять нашим пользователям данные высочайшего качества.
2100 Биоанализатор Agilent представляет собой основанную на микрофлюидике платформу, которая обеспечивает точное определение размера, количественный анализ и контроль качества библиотек ДНК, РНК или NGS.Качество РНК определяется номером целостности РНК (RIN). Чем выше RIN, тем выше качество.
4200 Tapestation — это высокопроизводительный прибор для контроля качества, который позволяет анализировать фрагменты образцов РНК и ДНК (включая гДНК).
Флуорометр Qubit Fluorometer компании Invitrogen представляет собой флуориметрическую платформу и является лучшим выбором для количественного анализа ДНК и РНК. Qubit более точен, чем Nanodrop.
Qubit более точен, чем Nanodrop.
Прибор QuantaBio Q для количественной ПЦР используется для количественного определения библиотеки NGS и анализа экспрессии генов.
Экстракция ДНК, РНК:
Нет времени выделять РНК/ДНК из образцов? Без проблем. Мы сотрудничаем с биорепозиторием геномной медицины в Институте геномной медицины, чтобы предоставить эту услугу нашим пользователям. Пожалуйста свяжитесь с нами для помощи.
Хроматин/ДНК с самообслуживанием
Сфокусированный ультразвуковой анализатор Covaris S220 — это мощная система, использующая управляемую адаптивно-сфокусированную акустическую (АФА) ультразвуковую технологию для разрезания ДНК, РНК или хроматина на фрагменты определенной длины в образцах объемом от 25 мкл до 10 мл.Система также может быть использована для ультразвуковой обработки тканей. Пожалуйста, свяжитесь с нами, чтобы узнать о самообслуживании Covaris S220.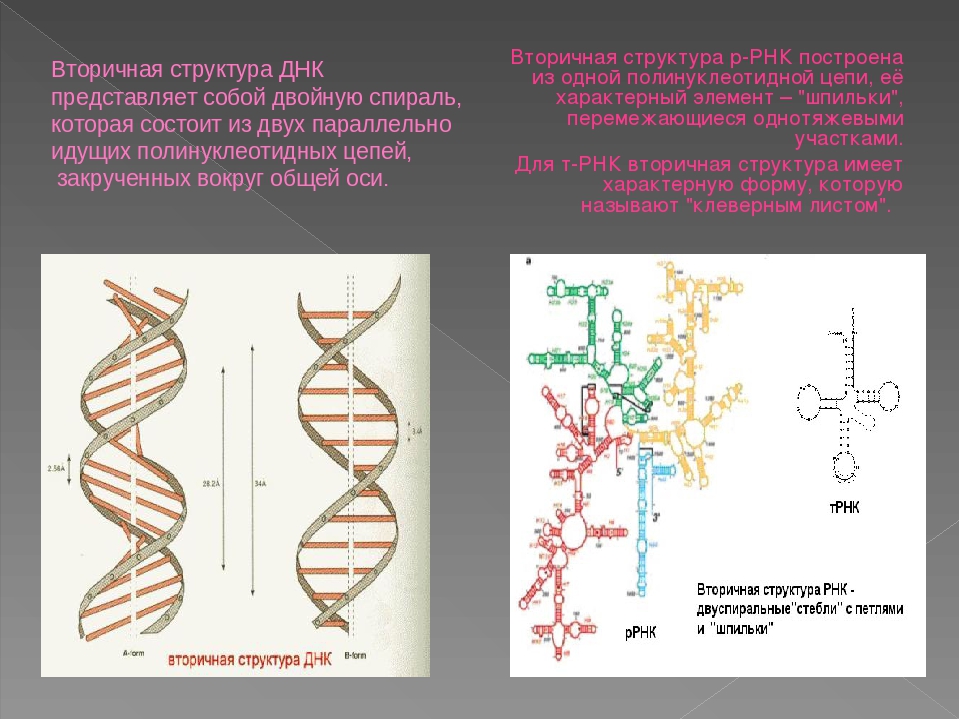
Секвенирование отдельных клеток
Для секвенирования отдельных клеток ядро Genomics Core и ядро проточной цитометрии работают как одна команда, обеспечивая наиболее эффективный и действенный рабочий процесс.
В ядре проточной цитометрии используется контроллер 10X Chromium для выделения клеток в отдельные секции. Затем контроллер Chromium может выполнять множество различных типов подготовки библиотек секвенирования следующего поколения, включая экспрессию генов отдельных клеток и другие протоколы, которые можно найти на веб-сайте 10X.После подготовки клеток и создания кДНК ядро проточной цитометрии передает отдельные клетки в ядро геномики.
Программа Genomics Core выполняет несколько проверок качества и дополнительные этапы подготовки библиотеки. Затем библиотеку можно секвенировать на NovaSeq 6000 или MiSeq, в зависимости от используемого протокола.
Анализ путей изобретательности (IPA)
Мы также предлагаем лицензионный доступ к следующему программному обеспечению Qiagen Ingenuity Analysis.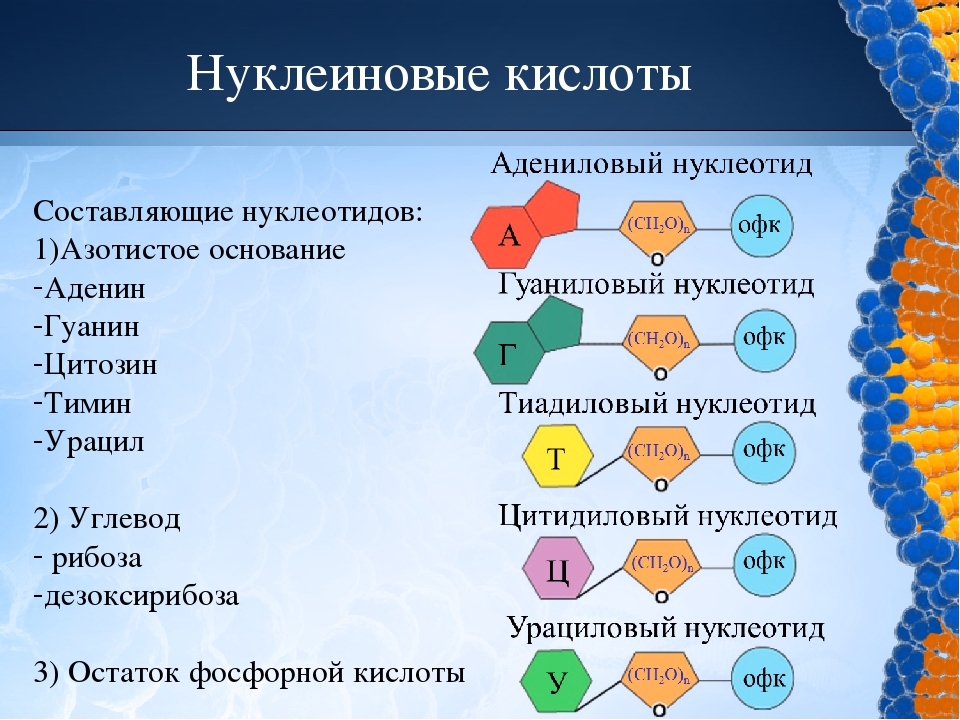 Дополнительную информацию можно найти на веб-сайте Qiagen Ingenuity. Пожалуйста, свяжитесь с нами, чтобы запросить доступ к лицензии IPA.
Дополнительную информацию можно найти на веб-сайте Qiagen Ingenuity. Пожалуйста, свяжитесь с нами, чтобы запросить доступ к лицензии IPA.
Услуги по Сэнгеру/капиллярному секвенированию
Компания Genomics Core сотрудничает с компаниями Eurofins и Genewiz в области капиллярного секвенирования и синтеза праймеров/олигонуклеотидов. Образцы секвенирования можно сбросить с погрузочной площадки и в коридоре NN возле почтовых ящиков. Образцы забираются в 15:30 с понедельника по пятницу, а результаты будут возвращены на ваш счет Eurofins на следующий день.Если у вас есть какие-либо вопросы, пожалуйста, свяжитесь с нами.
Оборудование
НоваСек 6000 NovaSeq 6000 — это мощная и эффективная система секвенирования со сверхвысокой производительностью, которая поддерживает самый широкий спектр приложений NGS и размеров исследований. Система использует непревзойденное качество данных, используя проверенную химию Illumina для секвенирования путем синтеза (SBS). NovaSeq — это модульная система, которая позволяет генерировать массивно-параллельные данные секвенирования с использованием множества различных проточных ячеек, необходимых для обработки пилотных и крупномасштабных геномных проектов.Посетите веб-сайт Illumina, чтобы ознакомиться со спецификациями разнообразных возможностей секвенирования.
NovaSeq — это модульная система, которая позволяет генерировать массивно-параллельные данные секвенирования с использованием множества различных проточных ячеек, необходимых для обработки пилотных и крупномасштабных геномных проектов.Посетите веб-сайт Illumina, чтобы ознакомиться со спецификациями разнообразных возможностей секвенирования.
Illumina MiSeq
Illumina MiSeq — это мощная настольная система секвенирования, подходящая для более специализированных приложений, таких как секвенирование ампликонов, секвенирование малых геномов, метагеномика, микроРНК-секвенирование, таргетная экспрессия генов и HLA-типирование, а также анализы рака и пользовательских генных панелей. Реагент MiSeq обеспечивает до 15 Гб выходных данных с 25 млн прочтений секвенирования и длинами прочтений 2 x 300 п.н. (фрагмент длиной 600 п.н.) и до 50 миллионов прочтений в зависимости от типа запуска.
- Каждый анализ MiSeq выполняется на однополосной проточной кювете.
 Образцы
Образцы - можно маркировать штрих-кодом и мультиплексировать для экономии средств.
- Длины считывания варьируются от 50, 75, 250 до 300 нуклеотидов.
- Секвенирование может выполняться в одном направлении (одностороннее считывание) или с обоих концов (парные концы).
Для секвенирования мы можем создать все библиотеки Illumina в ядре Genomics.Мы также принимаем библиотеки, подготовленные пользователями. Все библиотеки, в том числе представленные пользователями, будут проходить контроль качества за счет пользователя для обеспечения качества. Мы не несем ответственности за сбой секвенирования библиотек, отправленных пользователями, если это не вызвано поломкой секвенатора. Пожалуйста, предоставьте не менее 10 мкл объединенных библиотек с минимальной концентрацией 10 нМ (см. спецификации цикла ниже).
Длины чтения MiSeq и выходные данные :
- 1 x 75 nt /2 x 75 nt считываний PE (15/50 миллионов считываний/проточная кювета)
- 1 x 150 nt /2 x 150 nt считываний PE (10/30 миллионов считываний/проточная кювета)
- 1 x 250 нт/2 x 250 нт считываний PE (10/30 миллионов считываний/проточная кювета
- 1 x 300 nt /2 x 300 nt считываний PE (25/50 миллионов считываний/проточная кювета)
Доставка данных:
Ядро геномики отправит исследователям данные FastQ с информацией об обработке образцов. Данные для пользователей Cleveland Clinic будут храниться в соответствующих папках исследователей, доступных на общих дисках Genomics Core. Пользователи будут проинформированы по электронной почте после завершения проектов.
Данные для пользователей Cleveland Clinic будут храниться в соответствующих папках исследователей, доступных на общих дисках Genomics Core. Пользователи будут проинформированы по электронной почте после завершения проектов.
Outside Cleveland Clinic получат электронное письмо с информацией о том, как загрузить свои данные с нашего защищенного FTP-сервера.
Illumina iScan
- Чипы микрочипов сканируются с помощью Illumina iScan.
- Пользователи предоставляют массивы (человеческие или мышиные) и общую РНК. Core выполнит этап контроля качества РНК, обработает образцы РНК, если они пройдут, гибридизируют с массивами и создадут соответствующие выходные файлы (в Cleveland Clinic команда Core может установить программное обеспечение Illumina GenomeStudio на ПК в вашей лаборатории).
- Предоставляем бесплатные консультации по планированию эксперимента и подготовке образцов.

Биоанализатор Agilent и тапестация
Системы биоанализатора и тапестации Agilent позволяют проводить быстрый качественный анализ образцов РНК и ДНК.Автоматические системы электрофореза позволяют проводить точный анализ качества РНК и ДНК, начиная от разложившихся образцов и заканчивая малым количеством образцов.
Прибор для количественной ПЦР QuantaBio Q
Прибор QuantaBio Q для количественной ПЦР используется для количественного определения библиотеки NGS и анализа экспрессии генов.
Covaris S220 Фокусированный ультразвуковой датчик
Сфокусированный ультразвуковой анализатор Covaris S220 — это мощная система, использующая управляемую адаптивную сфокусированную акустическую (АФА) ультразвуковую технологию для разделения ДНК, РНК или хроматина на фрагменты определенной длины в образцах объемом от 25 мкл до 10 мл. Система также может быть использована для ультразвуковой обработки тканей. Пожалуйста, свяжитесь с нами, чтобы узнать о самообслуживании Covaris S220.
Система также может быть использована для ультразвуковой обработки тканей. Пожалуйста, свяжитесь с нами, чтобы узнать о самообслуживании Covaris S220.
Хромированный контроллер 10X
Core использует контроллер 10X Chromium для разделения ячеек на отдельные разделы. Контроллер Chromium затем может выполнять множество различных типов подготовки библиотеки секвенирования следующего поколения, включая экспрессию гена одной клетки и другие протоколы, которые можно найти на веб-сайте 10X.После подготовки клеток и создания кДНК ядро проточной цитометрии передает отдельные клетки в ядро геномики.
Информация о гранте
The Genomics Core предлагает широкий спектр услуг в области геномики, включая высокопроизводительное секвенирование, секвенирование РНК отдельных клеток, подготовку библиотек ДНК и РНК, приложения для микрочипов и генотипирования, а также контроль качества образцов. Мы оснащены системами Novaseq 6000 и MiSeq для различных приложений секвенирования, включая секвенирование РНК, секРНК, секвенирование микроРНК, ДНК-секвенирование, секвенирование всего экзома (WES), секвенирование всего генома (WGS), но не ограничиваясь ими. ), секвенирование ампликонов, микробная метагеномика и метатранскриптомика.В Genomics Core используется iScan Illumina для сканирования чипов генотипа/микрочипов и контроля качества образцов с использованием биоанализатора Agilent 2100, Tapestation 4200, флуорометра Qubit, количественной ПЦР и ультразвукового прибора Covaris S2. Кроме того, Genomics Core является центром для всех потребностей исследователей Кливлендской клиники в секвенировании внешних капилляров и синтезе олигонуклеотидов. Лицензия Ingenuity Pathway Analysis (IPA) поддерживается ядром геномики и доступна пользователям ядер геномики и/или протеомики.Genomics Core является общим ресурсом Комплексного онкологического центра NCI (P30CA043703-28) и NIH Cleveland Clinical and Translational Science Collaborative (CTSC).
Мы оснащены системами Novaseq 6000 и MiSeq для различных приложений секвенирования, включая секвенирование РНК, секРНК, секвенирование микроРНК, ДНК-секвенирование, секвенирование всего экзома (WES), секвенирование всего генома (WGS), но не ограничиваясь ими. ), секвенирование ампликонов, микробная метагеномика и метатранскриптомика.В Genomics Core используется iScan Illumina для сканирования чипов генотипа/микрочипов и контроля качества образцов с использованием биоанализатора Agilent 2100, Tapestation 4200, флуорометра Qubit, количественной ПЦР и ультразвукового прибора Covaris S2. Кроме того, Genomics Core является центром для всех потребностей исследователей Кливлендской клиники в секвенировании внешних капилляров и синтезе олигонуклеотидов. Лицензия Ingenuity Pathway Analysis (IPA) поддерживается ядром геномики и доступна пользователям ядер геномики и/или протеомики.Genomics Core является общим ресурсом Комплексного онкологического центра NCI (P30CA043703-28) и NIH Cleveland Clinical and Translational Science Collaborative (CTSC).
Несколько вариантов подготовки библиотеки, доступных для исследователей, заинтересованных в секвенировании РНК или ДНК, включая образцы с расщепленным или низкоконцентрированным геномным материалом. В сотрудничестве с ядром проточной цитометрии ядро геномики готовит библиотеки 10-кратных транскриптомов РНК отдельных клеток, что позволяет одновременно обрабатывать до десяти тысяч отдельных клеток на образец.Качество сконструированных геномных библиотек можно оценить с помощью микрожидкостных чипов, которые позволяют точно определить количество фрагментов РНК и ДНК. Геномные библиотеки последовательно секвенируются на одной из наших платформ Illumina HTS с возможностью предоставления от 300 Мб до 500 Гб данных и могут секвенировать фрагменты длиной до 300 п.н. (либо однонаправленные, либо парные концы). Персонал Genomics Core готов проконсультировать вас о том, какие варианты подготовки библиотеки и секвенирования лучше всего подходят для вашего эксперимента, в результате чего вы получите наиболее экономичные и надежные наборы данных, необходимые для уверенного поиска ответов на вопросы вашего исследования.
