
Урок 19. основные формы изменчивости организмов. генотипическая изменчивость — Биология — 9 класс
Изменчивость это способность организмов приобретать новые признаки под действием окружающей среды. Различают два вида изменчивости: генотипическую (наследственную) и фенотипическую (ненаследственную, или модификационную)
Изменчивость бывает:
1.Ненаследственная (фенотипическая)
2.Онтогенетическая (отражает закономерные изменения в ходе индивидуального развития организма)
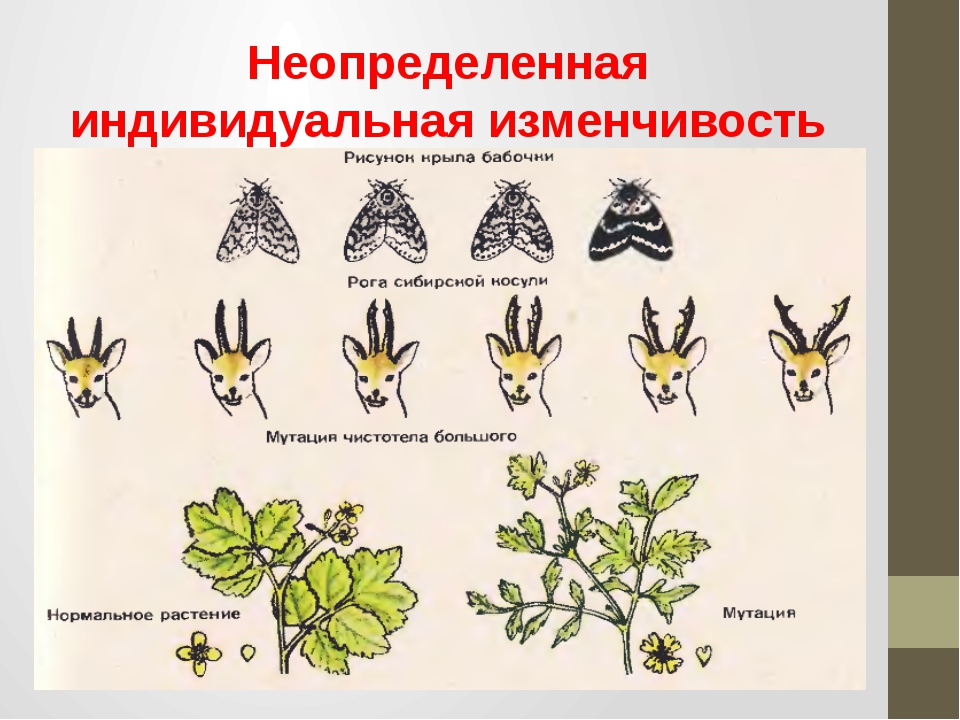
3.Наследственная (генотипическая, неопределенная, индивидуальная)
4.Соотносительная — коррелятивная, возникает в результате свойства генов влиять на формирование двух и более признаков(плейотропия).
Модификационная (ненаследственная) изменчивость
Модификационная изменчивость – изменение признаков и свойств организма, формирование фенотипа индивидуальной особи под влиянием генотипа и условий среды.
Мутационная изменчивость
Мутации – скачкообразные стойкие наследственные изменения структуры (качества) и количества ДНК данного организма, приводящие к изменению тех или иных признаков.
Основные положения мутационной теории де Фриза
(1901 – 1903 гг.)
1.Мутации – это дискретные изменения наследственного материала.
2.Мутации – редкие события.
3.Мутации могут устойчиво передаваться из поколения в поколение.
4.Мутации возникают спонтанно, ненаправленно.
5.Мутации могут быть вредными, полезными и нейтральными.
Мутации по месту возникновения делятся на соматические
возникают в клетках тела,
не передаются по наследству и генеративные, образуются в половых клетках, передаются по наследству.
по причинам возникновения могут быть спонтанные(естественные)
без вмешательства человека и индуцированные(искусственные)
вызываются мутагенами.

по локализации в клетке
-ядерные (идет
изменение хромосомного материала в ядре) и цитоплазматические ( идет изменение ДНК в митохондриях и хлоропластах)
по уровню возникновения : генные (точковые),
хромосомные (аберрации), геномные
Генные (точковые) мутации – (изменение нуклеотидной последовательности одного гена).
Существуют:
— Дупликации – повторение участка гена
— ВСТАВКИ – появление лишней пары нуклеотидов;
— ДЕЛЕЦИИ – выпадение нуклеотидов, замена нуклеотидных пар;
— ИНВЕРСИИ – переворот участка гена на 1800
Хромосомные мутации – аберрации (изменение структуры хромосом):
— ДЕФИШЕНСИ – потеря концевых участков хромосомы:
АБВГДЕ → БВГДЕ,
АБВГДЕ → АБВГД,
АБВГДЕ → БВГД;
— ДЕЛЕЦИЯ – выпадение участка хромосомы в средней части:
— ДУПЛИКАЦИЯ – удвоение участка хромосомы:
АБВГДЕ → АБВВГДЕ;
— ИНВЕРСИЯ – поворот участка хромосомы на 1800:
АБВГДЕ→АВБГДЕ;
— ТРАНСЛОКАЦИЯ – изменение положения участка хромосомы:
АБВГДЕ
АБВМНО
ИКЛМНО
ИКЛГДЕ
Геномные мутации (изменение числа хромосом в геноме):
1.
 Автополиплоидия – кратное увеличение гаплоидного набора хромосом в клетке (полиплоидия). Возникает при разрушении веретена деления, или выпадении цитокинеза при отсутствии редукционного деления во время мейоза. 4n, 6n.
Автополиплоидия – кратное увеличение гаплоидного набора хромосом в клетке (полиплоидия). Возникает при разрушении веретена деления, или выпадении цитокинеза при отсутствии редукционного деления во время мейоза. 4n, 6n.2. Аллополиплоидия – кратное увеличение числа хромосом у гибридов, полученных в результате скрещивания разных видов.
Тритикале – гибрид пшеницы и ржи
3. Гетероплоидия (анеуплоидия) – увеличение числа хромосом, не кратное гаплоидному.
2n + 1 – трисомик.
2n – 1 – моносомик.
2n – 2 – нуллисомик.
2n + х – полисомик.
Закон гомологических рядов
(Н.И. Вавилов, 1920 г.)
«Виды и роды, генетически близко связанные друг с другом единством происхождения, характеризуются сходными рядами наследственной изменчивости».
Зная, какие формы изменчивости встречаются у одного вида, можно предвидеть нахождение аналогичных форм у родственного ему вида.
Так, у разных классов позвоночных встречаются сходные мутации: альбинизм и отсутствие перьев у птиц; альбинизм и отсутствие шерсти у млекопитающих; гемофилия у многих млекопитающих и человека.

У растений наследственная изменчивость отмечена по таким признакам, как пленчатое или голое зерно, остистый или безостый колос.
Презентация по биологии «Наследственная изменчивость»
Наследственной изменчивостью обусловлены различные формы внутрипопуляционного полиморфизма. В некоторых популяциях наблюдается сосуществование двух или более ясно различимых форм (например, у двухточечной божьей коровки почти во всех популяциях встречаются черная форма с красными пятнами и красная форма с черными пятнами).
В основе этого явления могут лежать разные эволюционные механизмы: неодинаковая приспособленность сосуществующих форм к условиям различных сезонов года, повышенная жизнеспособность гетерозигот, в потомстве которых постоянно выщепляются обе гомозиготные формы или другие, еще недостаточно изученные механизмы.
Таким образом, и групповая, и индивидуальная изменчивости включают изменения как наследственной, так и ненаследственной природы.
Независимой изменчивости признаков противопоставляют коррелятивную изменчивость — взаимосвязанное изменение различных признаков и свойств: связь между ростом и весом особей (положительная корреляция) или темпом клеточного деления и величиной клеток (отрицательная корреляция).
Корреляции могут быть обусловлены чисто генетическими причинами (плейотропия) или взаимозависимостями процессов становления определенных признаков и свойств в индивидуальном развитии особей (онтогенетические корреляции), а также сходными реакциями разных признаков и свойств на одни и те же внешние воздействия (физиологические корреляции).
Наконец, корреляции могут отражать историю происхождения популяций из смеси двух или более форм, каждая из которых привносит не отдельные признаки, а комплексы взаимосвязанных признаков и свойств (исторические корреляции). Изучение коррелятивной изменчивости имеет важное значение в палеонтологии (например, при реконструкции вымерших форм по отдельным ископаемым остаткам), в антропологии (например, при восстановлении черт лица на основе изучения черепа), в селекции и медицине.
Изучение коррелятивной изменчивости имеет важное значение в палеонтологии (например, при реконструкции вымерших форм по отдельным ископаемым остаткам), в антропологии (например, при восстановлении черт лица на основе изучения черепа), в селекции и медицине.
Основные методы изучения изменчивостиь — сравнительно-описательный и биометрический.
Совокупность этих методов позволяет исследовать как паратипическую, так и генотипическую компоненты общей фенотипической изменчивости. Так, первую можно изучать, сравнивая генотипически идентичные клоны и чистые линии, развивающиеся в разных условиях.
Сложнее выделить чисто генотипическую изменчивость из общей фенотипической. Это возможно сделать на основе биометрического анализа. В медицинской генетике для тех же целей используется определение процента конкордантности (совпадения) тех или иных признаков у одно- и разнояйцевых близнецов.
Наследственность и изменчивость живых организмов иногда противопоставляют как «консервативное» и «прогрессивное» начала.
В действительности же они теснейшим образом связаны. Отсутствие полной стабильности генотипа обусловливает мутационную и (в ходе дальнейших скрещиваний и расщеплений) комбинационную изменчивость, то есть в целом — генотипическую изменчивость.
Паратипическая (ненаследственная) изменчивость — результат лишь относительной стабильности генотипа при определении им в онтогенезе нормы реакции при развитии признаков и свойств особей. Из этого следует возможность экспериментальных воздействий как на наследственную, так и на ненаследственную изменчивость.
Первую можно усилить воздействием мутагенных факторов (излучения, температура, химические вещества).
Размах и направление комбинационной изменчивости можно контролировать с помощью искусственного отбора. На ненаследственную изменчивость можно воздействовать, изменяя условия среды (питание, свет, влажность и т.д.), в которых протекает развитие организма.
Четкое представление о категориях и формах изменчивости необходимо при построении эволюционных схем и теорий, так как явления наследственности и изменчивости лежат в основе эволюционного процесса, а также в практической селекции растений и животных, при изучении ряда проблем медицинской географии и популяционной антропологии.
Индивидуальная изменчивость — это, что такое, какие, определение, значение, доклад, реферат, конспект, сообщение, вики — WikiWhat
Основная статья: ИзменчивостьСодержание (план)
Индивидуальная изменчивость — отличие организмов нового поколения любого растения и животного от родителей, а также друг от друга отдельными признаками и свойствами (рис. 27).
Основная причина изменчивости организмов связана с изменением окружающей среды — температуры, влажности, воздуха, пищи и других факторов. Влияние внешней среды на организм осуществляется в определённом и неопределённом виде. В первом случае воздействие внешней среды проявляется у всех организмов, а во втором — у отдельных организмов. Иначе говоря, в первом случае имеет место групповая изменчивость, во втором — индивидуальная.
Факторы внешней среды могут оказывать на организмы прямое или косвенное влияние. В результате прямого влияния внешней среды изменяется организм, а при косвенном влиянии изменяются его последующие поколения. Материал с сайта http://wikiwhat.ru
Материал с сайта http://wikiwhat.ru
«Сомнительные» виды
Существование индивидуальной изменчивости у организмов Дарвин доказал путём сопоставления вида с его разновидностями. Под «разновидностью вида» он понимал группу организмов с неярко выраженными признаками и свойствами, присущими данному виду. Промежуточные формы между двумя отдельными видами не встречаются. Однако наличие промежуточных форм между видом и его разновидностью совершенно естественно. Поэтому во времена Дарвина разновидности видов называли также «сомнительными» видами. Вследствие наличия в природе разновидностей видов учёные затрудняются в определении численности видов. Основная причина этого состоит в том, что одни учёные считают группу организмов, не достигших степени проявления признаков и свойств, видом, а другие — разновидностью вида. Во времена Дарвина во флоре Англии насчитывалось 182 «сомнительных» вида.
Картинки (фото, рисунки)
 27. Изменчивость растений, животных и бактерий (цветки хризантемы, початки кукурузы, бактерии, рога оленя, гребни кур, глаза мушки дрозофилы)» data-thumb=»http://wikiwhat.ru/public/page_images/574/0x65-27.jpg» data-src=»http://wikiwhat.ru/public/page_images/574/27.jpg»>
27. Изменчивость растений, животных и бактерий (цветки хризантемы, початки кукурузы, бактерии, рога оленя, гребни кур, глаза мушки дрозофилы)» data-thumb=»http://wikiwhat.ru/public/page_images/574/0x65-27.jpg» data-src=»http://wikiwhat.ru/public/page_images/574/27.jpg»>Рис. 27. Изменчивость растений, животных и бактерий (цветки хризантемы, початки кукурузы, бактерии, рога оленя, гребни кур, глаза мушки дрозофилы)
-
Что такое групповая и индивидуальная изменчивость?
-
Что такое определённое и неопределённое влияние внешней среды на организмы?
-
В чём проявляется прямое и косвенное влияние внешней среды на организмы?
-
Что такое «сомнительный» вид?
Изменчивость: наследственная и ненаследственная | Биология. Реферат, доклад, сообщение, краткое содержание, конспект, сочинение, ГДЗ, тест, книга
Вопрос 1. Какие виды изменчивости вам известны?
Какие виды изменчивости вам известны?
Существует два основных вида изменчивости — ненаследственная и наследственная.
Ненаследственная (фенотипическая или модификационная) изменчивость — это процесс появления новых признаков под влиянием факторов внешней среды, не затрагивающих генотип. В качестве примера можно привести дуб, листья которого в процессе развития приобрели разную площадь в зависимости от освещенности (маленькую — при яркой освещенности, большую — при слабой).
Наследственная изменчивость связана с изменениями генотипа; признаки и свойства, приобретенные вследствие этого, передаются следующим поколениям.
Существует два типа наследственной изменчивости — комбинативная и мутационная.
Комбинативная изменчивость заключается в появлении новых признаков в результате образования новых комбинаций генов родителей в генотипах потомков. Комбинативную изменчивость обеспечивают случайное расхождение гомологичных хромосом в мейозе, обмен участками гомологичных хромосом в профазе I мейоза, случайная встреча гамет при оплодотворении, случайный выбор родительских пар.
Мутационная изменчивость обусловлена изменениями генов и хромосом.
Вопрос 2. Что такое норма реакции?
Норма реакции (иначе — пределы модификационной изменчивости) — это пределы, в которых возможно изменение признака при определенном генотипе. Норма реакции может быть как очень широкой (вес человека), так и очень узкой (группа крови). Обычно узкой нормой реакции обладают признаки, обеспечивающие жизненно важные качества организма. Важно также то, что от родителей потомству передается не жестко запрограммированное значение того или иного признака, а его норма реакции.
Вопрос 3. Почему фенотипическая изменчивость не передается по наследству?
Фенотипическая изменчивость не затрагивает генотип, обеспечивая лишь то или иное проявление заложенных в нем признаков. Она обычно предсказуема и у разных особей одного вида проходит однонаправленно. Например, если пшеничное поле не получает достаточно влаги, то у всех его растений плохо формируется колос.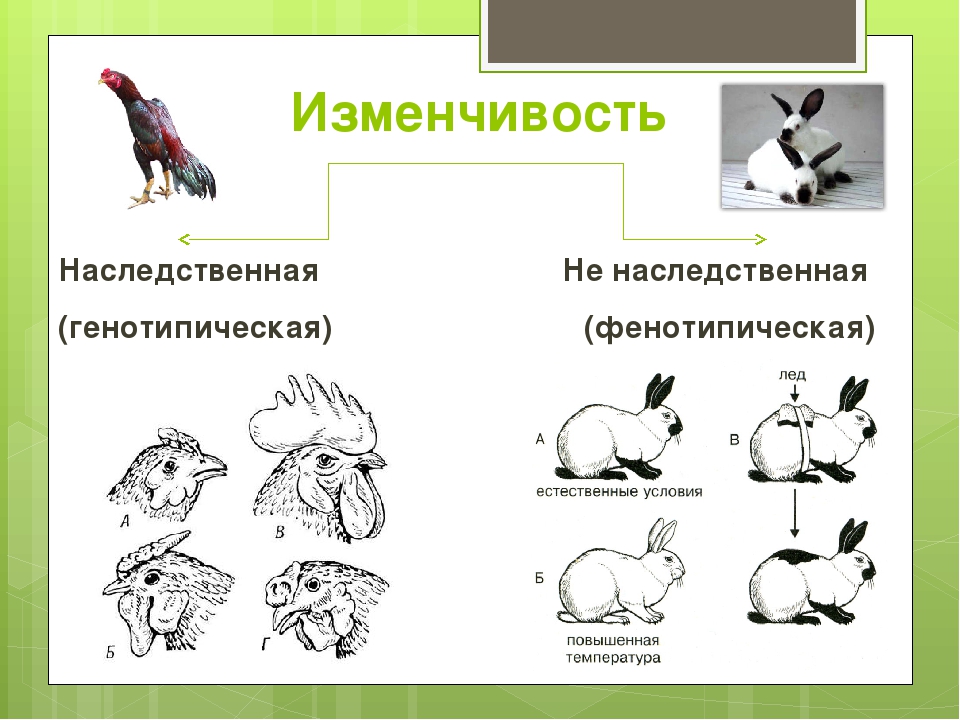 Генотип у особей в этом случае остается неизменным, поэтому передачи информации о модификациях потомству не происходит. Следовательно, фенотипическая изменчивость не наследуется.
Генотип у особей в этом случае остается неизменным, поэтому передачи информации о модификациях потомству не происходит. Следовательно, фенотипическая изменчивость не наследуется.
Вопрос 4. Что такое мутации? Охарактеризуйте основные свойства мутаций.
Мутации — это внезапные естественные или вызванные искусственно изменения генетического материала, приводящие к изменению тех или иных фенотипических признаков и свойств организма. Основные свойства мутаций:
- спонтанность — мутации возникают случайно;
- неспецифичность — могут возникать в любом участке генома;
- скачкообразность — вызывают новые качественные изменения;
- ненаправленность — возникшие изменения генотипа и фенотипа могут быть как биологически вредными, так и полезными.
Вопрос 5. Приведите классификацию мутаций по уровню изменений наследственного материала.
Различают три основных типа мутаций: Материал с сайта //iEssay. ru
ru
- генные мутации вызывают изменения в отдельных генах, нарушая порядок и число нуклеотидов в цепи ДНК. Это приводит к синтезу измененного (как правило, дефектного) белка. Следствием генных мутаций являются такие заболевания, как фенилкетонурия и мышечная дистрофия Дюшена;
- хромосомные мутации затрагивают значительный участок хромосомы, вызывая нарушения сразу в нескольких (иногда — многих) генах. Описаны случаи потери участка хромосомы, его переворота, перемещения, удвоения и т. п.;
- геномные мутации приводят к изменению числа хромосом в кариотипе. Они возникают в результате нарушения расхождения гомологичных хромосом. Примером может служить синдром Дауна, который возникает при появлении лишней 21-й хромосомы. При этом общее число хромосом становится равным 47. Другим примером геномных мутаций является формирование полиплоидных растений (чаще всего тетраплоидных).
Вопрос 6. Назовите основные группы мутагенных факторов.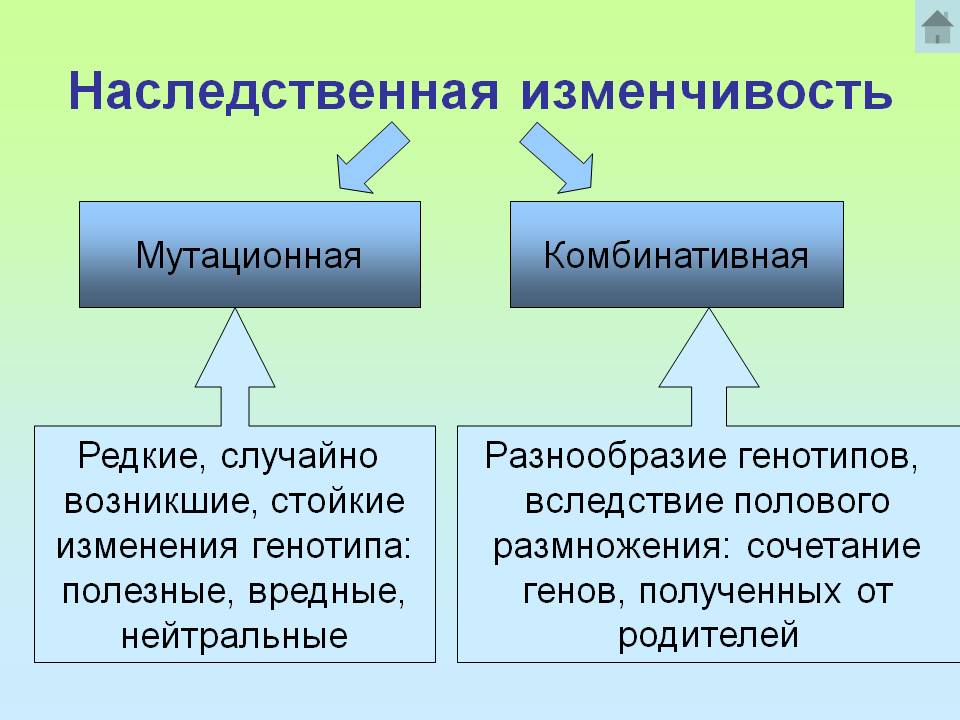 Приведите примеры мутагенов, относящихся к каждой группе.
Приведите примеры мутагенов, относящихся к каждой группе.
Мутагенные факторы можно разделить на три группы:
- физические мутагены — все типы ионизирующих излучений (у-лучи, рентгеновские лучи), ультрафиолетовое излучение, высокая и низкая температура;
- химические мутагены — аналоги нуклеиновых кислот, перекиси, соли тяжелых металлов (свинца, ртути), азотистая кислота, многие органические соединения;
- биологические мутагены — чужеродная ДНК и вирусы, которые, встраиваясь в ДНК хозяина, нарушают работу генов.
- ненаследственная изменчивость википедия
- готовые вопросы наследственная изменчивость
- наследственная изменчивость коротко
- фенотипическая изменчивость ненаследственная
- изменчивость наследственная и не наследственная
| Готовимся к самостоятельной работе по теме «Мутационная и модификационная изменчивость»!
| 8 тестов по теме45. Наследственная изменчивостьА) мутационная Б) определенная В) групповая Г) модификационная 136. Мутационная изменчивость, в отличие от модификационной, А) носит обратимый характер Б) передаётся по наследству В) характерна для всех особей вида Г) является проявлением нормы реакции признака 446. Значение мутационной изменчивости для эволюции, в отличие от модификационной изменчивости, состоит в том, что она А) возникает сразу у большого числа особей Б) возникает только у отдельных особей В) передаётся по наследству Г) не передаётся по наследству 529.  Мутационная изменчивость, в отличие от модификационной, Мутационная изменчивость, в отличие от модификационной,А) носит обратимый характер Б) передается по наследству В) носит массовый характер Г) не связана с изменением хромосом 809. Мутации отличаются от модификаций тем, что они А) сохраняются у потомков при отсутствии вызвавшего их фактора Б) возникают одновременно у многих особей в популяции В) всегда имеют адаптивный характер Г) обусловливают определенную изменчивость 926. Появление у человека загара является примером изменчивости А) комбинативной Б) мутационной В) генотипической Г) модификационной 1919. Изменчивость, сформировавшаяся как приспособленность к условиям внешней среды А) генотипическая Б) геномная В) индивидуальная Г) модификационная 2019. Какая изменчивость обеспечивает эволюцию видов? А) модификационная Б) возрастная В) генотипическая Г) географическая |

 Большинство мутаций – вредные. Мутации поставляют материал для естественного отбора, а уже ЕО приспосабливает организмы к условиям. Модификации – это приспособительная изменчивость, потому что они соответствуют окружающей среде.
Большинство мутаций – вредные. Мутации поставляют материал для естественного отбора, а уже ЕО приспосабливает организмы к условиям. Модификации – это приспособительная изменчивость, потому что они соответствуют окружающей среде.Оценка внутри- и межиндивидуальных вариаций транскриптома плаценты человека | Genome Biology
Из 159 миллионов полученных высококачественных прочтений 117 миллионов сопоставлены с аннотированными экзонами. В среднем было получено 1,46 миллиона прочтений с картированием экзонов для каждой библиотеки (репликация образца), что соответствует в среднем 2,9 миллиона прочтений с картированием экзонов для каждого человека (рисунок S1 в дополнительном файле 1). Было по крайней мере одно картированное чтение для каждой библиотеки в 13 156 генах, включая, помимо прочего, 11 301 белок-кодирующий ген, 801 псевдоген, 893 длинных некодирующих РНК (днРНК) и 40 малых РНК, включая 21 премиРНК.Уровни экспрессии нормализовали (стабилизировали дисперсию) с использованием протоколов, описанных в пакете DESeq2 [30]. Коэффициент корреляции Пирсона для каждой пары повторов выборки составлял 0,98 ± 0,005, что дает значение r-квадрата 0,96 ± 0,01. Качество данных было дополнительно оценено путем проверки профилей экспрессии трех генов с помощью RT-qPCR, среднее значение r Пирсона 0,74 ± 0,07 наблюдалось между значениями экспрессии, измеренными с помощью секвенирования РНК, по сравнению с RT-qPCR (рис. S2 в дополнительном файле 1).
В среднем было получено 1,46 миллиона прочтений с картированием экзонов для каждой библиотеки (репликация образца), что соответствует в среднем 2,9 миллиона прочтений с картированием экзонов для каждого человека (рисунок S1 в дополнительном файле 1). Было по крайней мере одно картированное чтение для каждой библиотеки в 13 156 генах, включая, помимо прочего, 11 301 белок-кодирующий ген, 801 псевдоген, 893 длинных некодирующих РНК (днРНК) и 40 малых РНК, включая 21 премиРНК.Уровни экспрессии нормализовали (стабилизировали дисперсию) с использованием протоколов, описанных в пакете DESeq2 [30]. Коэффициент корреляции Пирсона для каждой пары повторов выборки составлял 0,98 ± 0,005, что дает значение r-квадрата 0,96 ± 0,01. Качество данных было дополнительно оценено путем проверки профилей экспрессии трех генов с помощью RT-qPCR, среднее значение r Пирсона 0,74 ± 0,07 наблюдалось между значениями экспрессии, измеренными с помощью секвенирования РНК, по сравнению с RT-qPCR (рис. S2 в дополнительном файле 1). . Таким образом, основанные как на повторах образцов, так и на независимом методе измерения количества экспрессии, полученные нами данные обеспечивают точное измерение количества транскриптов РНК.
. Таким образом, основанные как на повторах образцов, так и на независимом методе измерения количества экспрессии, полученные нами данные обеспечивают точное измерение количества транскриптов РНК.
Полная структура экспрессии генов
Чтобы определить, была ли межиндивидуальная вариация экспрессии гена больше, чем внутрииндивидуальная вариация, и группировались ли индивидуумы по происхождению, была рассчитана матрица корреляции выборка за выборкой и построена иерархическая кластеризованная дендрограмма всех библиотек. был произведен (рис. 1A). Мы заметили, что 74 из 80 повторов вскрытия сгруппированы вместе, что согласуется с результатами корреляции и указывает на то, что внутрииндивидуальные вариации, как правило, меньше, чем межиндивидуальные вариации.Три особи, чьи повторы вскрытия не спарились, впоследствии были исключены из всех дальнейших анализов, исходя из предположения, что их отсутствие спаривания было результатом вскрытия и/или ошибки обработки.
Рисунок 1
Обзор общей изменчивости экспрессии генов в 13156 экспрессированных генах. (A) Дендрограмма кластера библиотек, основанная на следующем расстоянии экспрессии между каждой парой библиотек: 1-abs(r), где r — коэффициент корреляции Пирсона для уровней экспрессии всех генов.Отдельные библиотеки и филиалы окрашены в цвет, чтобы обозначить их групповую принадлежность; звездочки обозначают три пары библиотек реплик, которые не кластеризуются вместе. (B) Диаграммы рассеивания первых трех основных компонентов (PC) с использованием данных отдельных лиц и всех генов. Объясненная пропорция вариации аннотирована на каждой оси. (C) Графики рассеяния между первыми несколькими ПК и коррелированными независимыми переменными.
(A) Дендрограмма кластера библиотек, основанная на следующем расстоянии экспрессии между каждой парой библиотек: 1-abs(r), где r — коэффициент корреляции Пирсона для уровней экспрессии всех генов.Отдельные библиотеки и филиалы окрашены в цвет, чтобы обозначить их групповую принадлежность; звездочки обозначают три пары библиотек реплик, которые не кластеризуются вместе. (B) Диаграммы рассеивания первых трех основных компонентов (PC) с использованием данных отдельных лиц и всех генов. Объясненная пропорция вариации аннотирована на каждой оси. (C) Графики рассеяния между первыми несколькими ПК и коррелированными независимыми переменными.
Дополнительным наблюдением из дендрограммы корреляции выборки является отсутствие кластеризации лиц с одинаковым происхождением.Для дальнейшей оценки этого анализ основных компонентов (PC) показывает, что в отличие от того, что обычно наблюдается для генетических данных [31-33], в этом клеточном фенотипе нет очевидной структуры, соответствующей группам (рис. 1B). Однако, когда нагрузки PC для каждого человека проверяются на корреляцию с другими аспектами данных (рис. 1C), PC2 коррелирует с длиной плода при рождении (r = -0,54, значение Bonferroni P = 0,007), PC3 коррелирует с сумма сопоставленных чтений (r = -0.62, значение Bonferroni P = 0,0005), а PC4 коррелирует с нормальным весом матери (r = 0,46, значение Bonferroni P = 0,045). Кроме того, анализ генов, которые коррелируют с загрузками из первых трех ПК [34,35], обнаруживает обогащение в сотнях категорий генной онтологии, в частности, молекулярная функция (GO:0003674), биологический процесс (GO:0008150), связывание (GO: 0005488) и их подкатегории (Дополнительный файл 2: Таблица A), а также многочисленные пути KEGG (Дополнительный файл 2: Таблица B), выделенные наиболее обогащенным путем KEGG, а именно 01100: Метаболические пути (скорректированное значение P = 2.9е-05). В целом, оказывается, что общая изменчивость транскриптома в значительной степени зависит от факторов, отличных от групповой принадлежности (т.
1B). Однако, когда нагрузки PC для каждого человека проверяются на корреляцию с другими аспектами данных (рис. 1C), PC2 коррелирует с длиной плода при рождении (r = -0,54, значение Bonferroni P = 0,007), PC3 коррелирует с сумма сопоставленных чтений (r = -0.62, значение Bonferroni P = 0,0005), а PC4 коррелирует с нормальным весом матери (r = 0,46, значение Bonferroni P = 0,045). Кроме того, анализ генов, которые коррелируют с загрузками из первых трех ПК [34,35], обнаруживает обогащение в сотнях категорий генной онтологии, в частности, молекулярная функция (GO:0003674), биологический процесс (GO:0008150), связывание (GO: 0005488) и их подкатегории (Дополнительный файл 2: Таблица A), а также многочисленные пути KEGG (Дополнительный файл 2: Таблица B), выделенные наиболее обогащенным путем KEGG, а именно 01100: Метаболические пути (скорректированное значение P = 2.9е-05). В целом, оказывается, что общая изменчивость транскриптома в значительной степени зависит от факторов, отличных от групповой принадлежности (т. е. популяции), и, следовательно, изменчивость транскриптома не соответствует ожидаемым паттернам генетической структуры для этих групп [32,36].
е. популяции), и, следовательно, изменчивость транскриптома не соответствует ожидаемым паттернам генетической структуры для этих групп [32,36].
Распределение вариаций экспрессии генов
Суммарная дисперсия экспрессии каждого гена распределялась между группами (Mst и Nst), между особями внутри групп (Mit и Nit) и между репликами вскрытия (или внутри особей, Met и Net) .Анализ дисперсии (ANOVA) для каждого гена был выполнен для распределения вариации, и для получения оценок распределения использовались два компонента данных — аддитивные компоненты дисперсии и суммы квадратов оценок (см. раздел «Методы» для получения подробной информации об этих модели). В рамках этой структуры мы можем моделировать все группы одновременно, а также моделировать популяции попарно. Предполагая модель с четырьмя популяциями, параметры дисперсии (Mst, Mit, Met) и вариации (Nst, Nit, Net) сильно коррелируют между генами (Mst:Nst, r = 0.97; Mit:Nit, r = 0,95; Мет: чистая, r = 0,99; P = 2,2e-16; Рисунок S3 в дополнительном файле 1), хотя их распределения и средние оценки сильно различаются (рис. 2A–C). Уникальность параметров дисперсии (M*t) отражает специфический способ получения этих значений, то есть аддитивный компонент дисперсии от ожидаемых средних квадратов в этом иерархическом дисперсионном анализе типа I (см. Таблицы S1 и S2 в Дополнительных файл 1). Учитывая корреляцию между оценками параметров и отсутствие нулевых значений в приближении суммы квадратов (рис. S3 в дополнительном файле 1), мы сосредоточимся на параметрах вариации или изменчивости Nst, Nit и Net.В среднем мы находим, что 33,2% вариабельности экспрессии генов обнаруживаются среди популяций клеток в пределах одной ткани (Net, перестановка прочтений среди повторов, P = 0,22), 58,9% вариабельности приходится на особей внутри групп ( Nit, перестановка библиотек среди особей внутри групп, P = 0,048), и 7,9% изменчивости приходится на группы (Nst, перестановка особей среди групп, P = 0,24) (рис. 2B и C). Эти оценки показывают, что хотя межиндивидуальная изменчивость в среднем является самым большим компонентом изменчивости экспрессии, внутрииндивидуальная вариация не может быть проигнорирована при измерении клеточных фенотипов.
2A–C). Уникальность параметров дисперсии (M*t) отражает специфический способ получения этих значений, то есть аддитивный компонент дисперсии от ожидаемых средних квадратов в этом иерархическом дисперсионном анализе типа I (см. Таблицы S1 и S2 в Дополнительных файл 1). Учитывая корреляцию между оценками параметров и отсутствие нулевых значений в приближении суммы квадратов (рис. S3 в дополнительном файле 1), мы сосредоточимся на параметрах вариации или изменчивости Nst, Nit и Net.В среднем мы находим, что 33,2% вариабельности экспрессии генов обнаруживаются среди популяций клеток в пределах одной ткани (Net, перестановка прочтений среди повторов, P = 0,22), 58,9% вариабельности приходится на особей внутри групп ( Nit, перестановка библиотек среди особей внутри групп, P = 0,048), и 7,9% изменчивости приходится на группы (Nst, перестановка особей среди групп, P = 0,24) (рис. 2B и C). Эти оценки показывают, что хотя межиндивидуальная изменчивость в среднем является самым большим компонентом изменчивости экспрессии, внутрииндивидуальная вариация не может быть проигнорирована при измерении клеточных фенотипов. Сходным образом, в то время как среди групповых вариаций экспрессии в среднем не достигают уровней структуры, наблюдаемых на генетическом уровне, групповой компонент заметно влияет на вариации экспрессии, особенно в подмножестве генов, которые мы исследуем ниже.
Сходным образом, в то время как среди групповых вариаций экспрессии в среднем не достигают уровней структуры, наблюдаемых на генетическом уровне, групповой компонент заметно влияет на вариации экспрессии, особенно в подмножестве генов, которые мы исследуем ниже.
Сводка распределения. (A) Распределение долей дисперсии, полученных из аддитивного компонента оценок дисперсии. (B) Распределение вариационных долей, полученных из суммы квадратов. (C) Средние оценки для каждого параметра распределения с использованием как дисперсии, так и вариации. (D) Дендрограмма средневзвешенных расстояний населения, полученная из параметра Mst. (E) Дендрограмма средневзвешенных расстояний населения, полученная из параметра Nst.
При попарном моделировании вариации экспрессии средние оценки аналогичны тем, которые наблюдались при анализе четырех популяций (таблица 1). Однако внутригрупповая изменчивость (Nst) находится в пределах 0.045 (для AF:EU) до 0,062 (для EA:SA). Дендрограмма была построена с использованием средних попарных расстояний Nst (рис. 2D и E). Мы находим, что данные совпадают с ожиданиями от генетических данных [36], за исключением того, что SA, как правило, является самой отдаленной группой.
Однако внутригрупповая изменчивость (Nst) находится в пределах 0.045 (для AF:EU) до 0,062 (для EA:SA). Дендрограмма была построена с использованием средних попарных расстояний Nst (рис. 2D и E). Мы находим, что данные совпадают с ожиданиями от генетических данных [36], за исключением того, что SA, как правило, является самой отдаленной группой.
Средняя экспрессия и оценки распределения
Средняя экспрессия каждого гена значимо коррелирует с оценкой остаточной (или внутрииндивидуальной) суммы квадратов (Pearson r = 0.60, P <0,001). Это показывает, что по мере увеличения средней экспрессии также увеличивается вариация содержания мРНК среди повторений нашего образца. Таким образом, мы оцениваем, что среднее выражение объясняет 36% вариации нашей суммы квадратов ошибок. Однако суммы квадратов среди групп (r = 0,018, P = 0,034) и среди особей внутри групп (r = -0,029, P = 0,001) слабее коррелируют со средним выражением. Следовательно, параметры распределения коррелируют со средним выражением с коэффициентами -0.446, 0,388 и 0,166 для Net, Nit и Nst ( P <0,001) соответственно. Таким образом, доля вариации, объясняемая средним выражением для каждого параметра распределения, составляет 20%, 15% и 2,7% для Net, Nit и Nst соответственно. Это говорит о том, что среднее выражение оказывает умеренное влияние на оценки параметров, и получение большего количества прочтений не будет сильно влиять на оценки распределения.
Следовательно, параметры распределения коррелируют со средним выражением с коэффициентами -0.446, 0,388 и 0,166 для Net, Nit и Nst ( P <0,001) соответственно. Таким образом, доля вариации, объясняемая средним выражением для каждого параметра распределения, составляет 20%, 15% и 2,7% для Net, Nit и Nst соответственно. Это говорит о том, что среднее выражение оказывает умеренное влияние на оценки параметров, и получение большего количества прочтений не будет сильно влиять на оценки распределения.
Дифференциальная экспрессия генов среди людей
Доля генов, которые значительно различаются по уровням экспрессии среди людей, была проанализирована с помощью теста F-отношения между индивидуальной и внутрииндивидуальной дисперсией.Мы заметили, что 5880 генов, или 44,5% всех генов (при FDR 5%), проявляли значительную изменчивость среди индивидуумов внутри группы. Кроме того, подгонка двух линейных моделей к данным (нулевая модель и вторая модель, которая включает индивидуумов в качестве объясняющей переменной) с последующим критерием подгонки модели методом хи-квадрат приводит к 5491 гену (41,7% всех генов) со значительными межиндивидуальная дисперсия (при FDR 5%). В обоих анализах значимые гены перекрываются на 84%. Мы оценили долю внутригрупповой изменчивости, объясняемой межиндивидуальной изменчивостью, с помощью параметра Nis (SSb/SSb + SSe; см. Методы).В среднем 64% внутригрупповой изменчивости приписывается отдельным лицам, что указывает на существенную межиндивидуальную изменчивость. Те гены, которые значительно дифференцированно экспрессируются (DE) среди людей, как определено с помощью теста F-ratio, имеют минимальное значение Nis 0,65. Чтобы определить, может ли быть значительная вариация, связанная с внутрииндивидуальной изменчивостью в некоторых локусах, мы инвертировали критерий F-отношения, поместив внутрииндивидуальные средние квадраты в числителе, а межиндивидуальные средние квадраты в знаменателе, но не наблюдали значительных изменений. loci после коррекции Бенджамини-Хохберга.В целом, это показывает, что существуют существенные межиндивидуальные различия в вариациях экспрессии генов.
В обоих анализах значимые гены перекрываются на 84%. Мы оценили долю внутригрупповой изменчивости, объясняемой межиндивидуальной изменчивостью, с помощью параметра Nis (SSb/SSb + SSe; см. Методы).В среднем 64% внутригрупповой изменчивости приписывается отдельным лицам, что указывает на существенную межиндивидуальную изменчивость. Те гены, которые значительно дифференцированно экспрессируются (DE) среди людей, как определено с помощью теста F-ratio, имеют минимальное значение Nis 0,65. Чтобы определить, может ли быть значительная вариация, связанная с внутрииндивидуальной изменчивостью в некоторых локусах, мы инвертировали критерий F-отношения, поместив внутрииндивидуальные средние квадраты в числителе, а межиндивидуальные средние квадраты в знаменателе, но не наблюдали значительных изменений. loci после коррекции Бенджамини-Хохберга.В целом, это показывает, что существуют существенные межиндивидуальные различия в вариациях экспрессии генов.
Дифференциальная экспрессия генов среди групп
Для идентификации и количественного определения генов, которые могут по-разному экспрессироваться среди групп людей, использовались три разных метода: два опубликованных метода (DESeq [30] и tweeDESeq [37]) и перестановка иерархического дисперсионного анализа. . Два опубликованных метода могут одновременно сравнивать только две группы, в то время как перестановки иерархического ANOVA позволяют анализировать две или более групп одновременно.
. Два опубликованных метода могут одновременно сравнивать только две группы, в то время как перестановки иерархического ANOVA позволяют анализировать две или более групп одновременно.
Несмотря на заметные различия в количестве генов DE, идентифицированных каждым методом, существуют устойчивые тенденции (таблица 2). Например, относительная доля генов DE для каждой пары популяций коррелировала между методами (Pearson r = 0,927, P <0,008), а сравнения, включавшие выходцев из Южной Азии, как правило, имели наибольшее количество генов DE для любой группы. Кроме того, 99% и 92% генов, идентифицированных как DE методами DESeq и tweeDESeq соответственно, также были идентифицированы как DE методом перестановки.В анализе перестановок пороговое значение Nst для генов DE немного различается в зависимости от сравниваемых групп, но в среднем составляет оценку Nst не менее 0,326. Уменьшенное количество генов DE, идентифицированных с помощью методов DESeq и tweeDESeq, связано с тем, что оба метода представляют собой анализ на основе моделей со специфическими тестами и коррекцией ложного обнаружения дифференциальной экспрессии. Представленный здесь метод перестановки просто идентифицирует экстремумы в наблюдаемых данных, которые трудно объяснить случайностью.
Представленный здесь метод перестановки просто идентифицирует экстремумы в наблюдаемых данных, которые трудно объяснить случайностью.
Чтобы определить потенциальную биологическую значимость генов, идентифицированных как DE, мы проверили их обогащение в путях GO и KEGG. При тестировании объединения всех генов DE с попарной перестановкой (1784 гена DE) мы наблюдали обогащение 15 путями KEGG и 371 GO-категорией при FDR средней достоверности 20% (5 KEGG и 201 GO при FDR высокой достоверности 5). %) (таблица 3, дополнительный файл 3: таблица A).В целом, обогащение KEGG и GO указывает на то, что гены, участвующие в клеточной передаче сигналов, иммунном ответе, развитии тканей и органов и путях метаболизма, являются DE среди групп.
Таблица 3 Обогащенные GO и KEGG пути для попарного объединения генов DEНенейтральные профили экспрессии генов
Хотя трудно определить, развивается ли экспрессия конкретного гена в соответствии с нейтральностью или в условиях отбора, мы можем идентифицировать профили экспрессии, которые соответствуют четырем конкретным моделям отбора: направленному, балансирующему, стабилизация и диверсификация. Важно отметить, что эти анализы не проверяют отклонения от нейтральности, а скорее идентифицируют гены, которые демонстрируют профили экспрессии, соответствующие отбору по количественным признакам [38,39]. Ожидается, что признаки при направленном отборе будут демонстрировать сдвиги в средней экспрессии среди групп, что выражается в большей изменчивости внутри группы по сравнению с изменчивостью внутри группы, и, следовательно, будут согласовываться с ранее идентифицированными генами DE. Примером уравновешивающего отбора является высокое разнообразие или изменчивость среди особей в популяции, но низкая изменчивость среди популяций.Стабилизирующий отбор приводит к низким уровням вариабельности экспрессии среди особей, в то время как диверсифицирующий отбор отражается в высоких уровнях вариабельности экспрессии среди особей. Мы идентифицировали гены, которые типизируют каждый профиль отбора, используя распределение оценок вариаций, оценок общей дисперсии экспрессии и ряд перестановок, как описано в разделе «Методы».
Важно отметить, что эти анализы не проверяют отклонения от нейтральности, а скорее идентифицируют гены, которые демонстрируют профили экспрессии, соответствующие отбору по количественным признакам [38,39]. Ожидается, что признаки при направленном отборе будут демонстрировать сдвиги в средней экспрессии среди групп, что выражается в большей изменчивости внутри группы по сравнению с изменчивостью внутри группы, и, следовательно, будут согласовываться с ранее идентифицированными генами DE. Примером уравновешивающего отбора является высокое разнообразие или изменчивость среди особей в популяции, но низкая изменчивость среди популяций.Стабилизирующий отбор приводит к низким уровням вариабельности экспрессии среди особей, в то время как диверсифицирующий отбор отражается в высоких уровнях вариабельности экспрессии среди особей. Мы идентифицировали гены, которые типизируют каждый профиль отбора, используя распределение оценок вариаций, оценок общей дисперсии экспрессии и ряд перестановок, как описано в разделе «Методы».
Используя данные модели, подходящей одновременно для всех четырех групп, мы наблюдаем, что вариация среди групп (log(SS a )) положительно коррелирует с вариацией среди индивидуумов (log(SS b ), Пирсон r = 0.579, P <2.2e-16), что согласуется с ожиданиями при нейтральности [40]. Кроме того, вариации внутри индивидуумов (log(SS e ) также положительно коррелируют с вариациями среди индивидуумов (r = 0,46 Пирсона, P <2,2e-16) и вариациями между группами (r Пирсона = 0,25, P <2,2e-16) (рис. 3A). Чтобы оценить долю транскриптома плаценты человека, которая может соответствовать нейтральным и ненейтральным ожиданиям, мы выполнили серию перестановок (см. Методы).По нашим оценкам, 64,8% всех генов согласуются с моделью нейтрального дрейфа для количественного признака [38]. Наиболее распространенным ненейтральным профилем изменчивости экспрессии генов является стабилизирующий отбор, который влияет примерно на 26% всех генов, за которым следуют направленный (646 генов, 4,9%), диверсифицирующий (635 генов, 4,8%) и уравновешивающий (173 гена) отбор. , 1,3%) селекция (рис. 3B; список всех генов см. в дополнительном файле 4).
, 1,3%) селекция (рис. 3B; список всех генов см. в дополнительном файле 4).
Оценка нейтрального и нейтральногоненейтральная эволюция плацентарного транскриптома человека. (A) Диаграмма рассеяния среди групповых и среди индивидуальных вариаций, измеренная по логарифму соответствующей суммы квадратов. Гены, которые были идентифицированы как имеющие модели изменчивости, соответствующие нейтральности или направленному, диверсифицирующему, стабилизирующему или уравновешивающему отбору, имеют цветовую кодировку. (B) Круговая диаграмма, показывающая долю генов, соответствующую определенному способу эволюции.
Когда каждый из этих способов отбора сопоставляется с распределением внутригрупповых и внутригрупповых вариаций (рис. 3A), мы можем определить почти дискретные участки распределения, которые отражают эти наблюдения.Интересно, что есть области распространения, где эти способы отбора перекрываются (рис.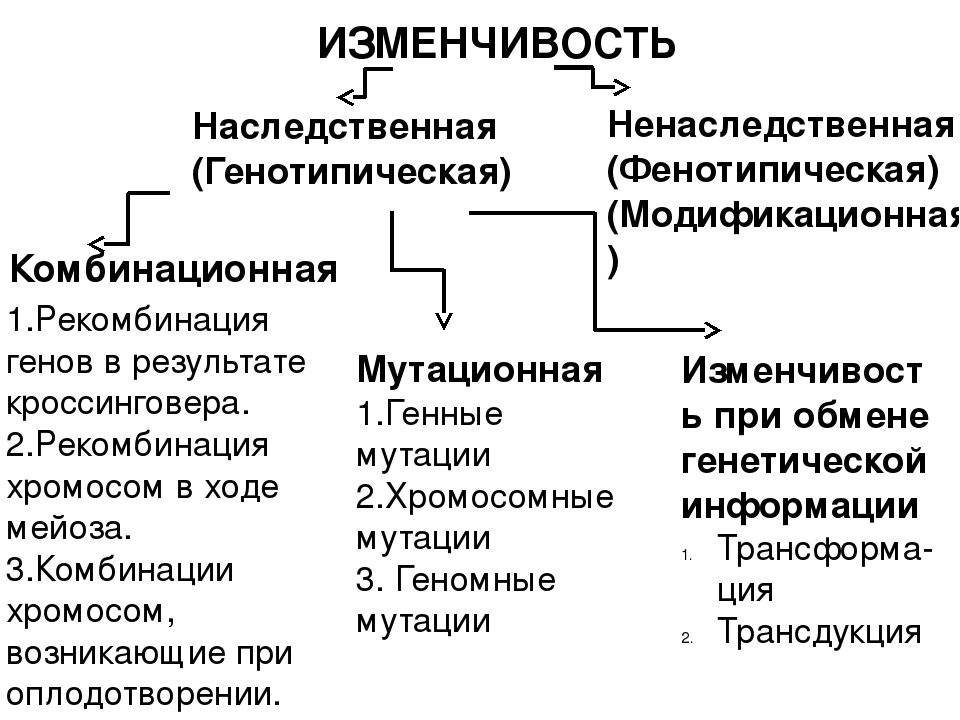 3B). Например, существует небольшой набор генов, для которых вариации экспрессии велики как среди отдельных людей (диверсифицирующие), так и среди групп (направленные) (рис. 4A и B). И наоборот, некоторые гены имеют большее ограничение в общей дисперсии, что соответствует стабилизирующему отбору, но также имеют значительные сдвиги в средней экспрессии среди групп, что соответствует направленному отбору (рис. 4A и C).И, наконец, ограниченное межиндивидуальное выражение (стабилизирующий отбор) также может происходить с уменьшением межгрупповой изменчивости (уравновешивающий отбор) (рис. 4D).
3B). Например, существует небольшой набор генов, для которых вариации экспрессии велики как среди отдельных людей (диверсифицирующие), так и среди групп (направленные) (рис. 4A и B). И наоборот, некоторые гены имеют большее ограничение в общей дисперсии, что соответствует стабилизирующему отбору, но также имеют значительные сдвиги в средней экспрессии среди групп, что соответствует направленному отбору (рис. 4A и C).И, наконец, ограниченное межиндивидуальное выражение (стабилизирующий отбор) также может происходить с уменьшением межгрупповой изменчивости (уравновешивающий отбор) (рис. 4D).
Блок-диаграммы вариации ненейтрального выражения. Ось Y всех графиков иллюстрирует один и тот же диапазон экспрессии. Каждая популяция имеет цветовую кодировку, а оценочное значение Nst для каждого гена находится в нижнем левом углу каждого графика. (A) Ген, соответствующий направленному отбору. (B) Ген, совместимый как с направленным, так и с диверсифицирующим отбором. (C) Ген, согласующийся как со стабилизирующим, так и с направленным отбором, с серой пунктирной горизонтальной линией, помогающей увидеть сдвиг в средней экспрессии, а также представленный ограниченно среди группы в пределах индивидуальной вариации. (D) Ген, соответствующий как стабилизирующему, так и уравновешивающему отбору.
(C) Ген, согласующийся как со стабилизирующим, так и с направленным отбором, с серой пунктирной горизонтальной линией, помогающей увидеть сдвиг в средней экспрессии, а также представленный ограниченно среди группы в пределах индивидуальной вариации. (D) Ген, соответствующий как стабилизирующему, так и уравновешивающему отбору.
Чтобы определить, могут ли гены, по-разному экспрессирующиеся среди групп, то есть гены с паттерном, согласующимся с направленным отбором, эффективно воспроизводить групповое происхождение, мы использовали вариации экспрессии во всех 646 направленных генах (тех, которые были идентифицированы при моделировании всех четырех популяций одновременно) для получения дерево UPGMA и выполнить анализ основных компонентов.Мы наблюдаем, что люди образуют монофилетические клады, соответствующие происхождению популяции (рис. 5A). Кроме того, при анализе основных компонентов наблюдались повышенные уровни структуры популяции, но они полностью различимы только при совместном просмотре первых трех ПК (рис. 5B). PC1 имеет тенденцию отличать лиц африканского происхождения от лиц неафриканского происхождения, в то время как PC2 имеет тенденцию отличать SA от EA, а PC3 отличает европейцев от неевропейцев (рисунок S4 в дополнительном файле 1).
5B). PC1 имеет тенденцию отличать лиц африканского происхождения от лиц неафриканского происхождения, в то время как PC2 имеет тенденцию отличать SA от EA, а PC3 отличает европейцев от неевропейцев (рисунок S4 в дополнительном файле 1).
Структура популяции, выявленная по генам, соответствующим направленному отбору. (A) Дерево экспрессии UPGMA расстояний между всеми библиотеками и индивидуумами в генах, согласующихся с направленным отбором. (B) Трехмерная диаграмма рассеяния первых трех ПК, основанная на вариациях в 646 генах, соответствующих направленному отбору. Доля объясненной вариации указана на каждой оси, а групповая принадлежность каждого человека имеет цветовую кодировку, чтобы соответствовать аннотации в (A) .
Изменчивость экспрессии, генетическое разнообразие и сетевая связность
Преобладание генов, которые отклоняются от ожиданий нейтрального дрейфа, особенно тех, которые согласуются со стабилизирующим отбором, побудило нас выдвинуть гипотезу о том, что межиндивидуальная изменчивость в экспрессии генов должна иметь генетический компонент. В частности, мы предположили, что гены с большим ограничением экспрессии будут иметь большее генетическое ограничение. Кроме того, гены, демонстрирующие большие межиндивидуальные вариации экспрессии, могут допускать, посредством смягчения ограничений или по необходимости, относительный избыток вариаций.Чтобы оценить эту гипотезу, мы проверили корреляцию между дисперсией экспрессии и попарным генетическим разнообразием. Попарное генетическое разнообразие (π) было рассчитано для каждого гена с учетом длины гена [41] для трех популяций по данным 1000 Genomes: CEU = североевропейцы, ASW = афроамериканцы с юго-запада США и CHS = ханьцы из южной части США. Китай. Мы выбрали эти три популяции, поскольку они являются лучшими доступными прокси для наших выборочных лиц. Когда разнообразие каждой популяции сравнивается с дисперсией экспрессии, мы наблюдаем значительную положительную корреляцию (ASW: r = 0.213; CEU: r = 0,189; CHS: r = 0,177, P < 2,2e-16 Рисунок S5 в дополнительном файле 1).
В частности, мы предположили, что гены с большим ограничением экспрессии будут иметь большее генетическое ограничение. Кроме того, гены, демонстрирующие большие межиндивидуальные вариации экспрессии, могут допускать, посредством смягчения ограничений или по необходимости, относительный избыток вариаций.Чтобы оценить эту гипотезу, мы проверили корреляцию между дисперсией экспрессии и попарным генетическим разнообразием. Попарное генетическое разнообразие (π) было рассчитано для каждого гена с учетом длины гена [41] для трех популяций по данным 1000 Genomes: CEU = североевропейцы, ASW = афроамериканцы с юго-запада США и CHS = ханьцы из южной части США. Китай. Мы выбрали эти три популяции, поскольку они являются лучшими доступными прокси для наших выборочных лиц. Когда разнообразие каждой популяции сравнивается с дисперсией экспрессии, мы наблюдаем значительную положительную корреляцию (ASW: r = 0.213; CEU: r = 0,189; CHS: r = 0,177, P < 2,2e-16 Рисунок S5 в дополнительном файле 1). Кроме того, дисперсия выражения также коррелирует со значениями D Tajima (ASW: r = 0,179; CEU: r = 0,129; CHS: r = 0,132, P < 2,2e-16. Эти наблюдения показывают, что общая дисперсия выражения имеет небольшую ( r-squared = 0,04), хотя и значительный генетический и, следовательно, наследуемый компонент
Кроме того, дисперсия выражения также коррелирует со значениями D Tajima (ASW: r = 0,179; CEU: r = 0,129; CHS: r = 0,132, P < 2,2e-16. Эти наблюдения показывают, что общая дисперсия выражения имеет небольшую ( r-squared = 0,04), хотя и значительный генетический и, следовательно, наследуемый компонент
Другим фактором, который может влиять на вариабельность экспрессии, является количество взаимодействующих партнеров гена.Предыдущая работа над генными сетями показала, что степень связности (количество взаимодействий) влияет на скорость молекулярной эволюции [42]. Здесь, используя данные из BioGrid, мы проверили, влияет ли количество взаимодействующих генов на дисперсию экспрессии гена (рисунок S6 в дополнительном файле 1). Действительно, мы наблюдаем слабую тенденцию к увеличению дисперсии экспрессии по мере уменьшения количества взаимодействующих генов (Pearson r = -0,28, P < 2,2e-16).
Чтобы оценить, как генетическое разнообразие и связность могут вместе влиять на изменчивость экспрессии генов, мы построили модель ANOVA, установив коэффициент вариации экспрессии генов в качестве переменной отклика и задав разнообразие и связность генов в качестве независимых переменных с взаимодействием. Каждый компонент модели значительно влиял на дисперсию экспрессии (разнообразие P < 2,2e-16; связность P < 2,2e-16; взаимодействие P = 0,029), объясняя примерно 4,3%, 2,3% и 0,07% общая дисперсия дисперсии выражений соответственно.
Каждый компонент модели значительно влиял на дисперсию экспрессии (разнообразие P < 2,2e-16; связность P < 2,2e-16; взаимодействие P = 0,029), объясняя примерно 4,3%, 2,3% и 0,07% общая дисперсия дисперсии выражений соответственно.
Модули коэкспрессии генов и функциональность категорий отбора
Чтобы определить, имеют ли наборы генов, соответствующие четырем ненейтральным способам эволюции, когерентный биологический эффект, мы проверили доказательства сетей коэкспрессии и обогащения GO термины генной онтологии и функциональные пути KEGG.Не наблюдалось обогащения генов, согласующихся с моделью уравновешивающего отбора. Результаты для трех других ненейтральных режимов представлены ниже.
В целом, гены, согласующиеся с направленным отбором (646 генов), были обогащены 145 категориями GO и шестью путями KEGG при FDR 20% (70 и 0 соответственно при FDR 5%). Они связаны с внеклеточными и мембранными областями, реакцией на стресс, инфекционное заболевание, путями и категориями передачи сигналов, связывания и метаболизма (дополнительный файл 3: таблица B). Было идентифицировано шесть модулей коэкспрессии, которые образуют компактные сети коэкспрессии, но также взаимодействуют друг с другом через уменьшенное количество локусов (рис. 6A и B). Единственный отдельный модуль, обогащенный определенным набором функций, — это модуль 6 (красный модуль на рис. 6A). Это самый маленький модуль, содержащий всего 54 гена, но при FDR 20% этот модуль обогащен 110 категориями GO (52 при FDR 5%, дополнительный файл 3: таблица C) и 15 путями KEGG (7 при FDR 5). %, Дополнительный файл 3: Таблица D).Эти гены в основном участвуют в защите и иммунном ответе, но также связаны с усвоением и перевариванием витаминов и метаболизмом арахидоновой кислоты, ключевой жирной кислоты.
Было идентифицировано шесть модулей коэкспрессии, которые образуют компактные сети коэкспрессии, но также взаимодействуют друг с другом через уменьшенное количество локусов (рис. 6A и B). Единственный отдельный модуль, обогащенный определенным набором функций, — это модуль 6 (красный модуль на рис. 6A). Это самый маленький модуль, содержащий всего 54 гена, но при FDR 20% этот модуль обогащен 110 категориями GO (52 при FDR 5%, дополнительный файл 3: таблица C) и 15 путями KEGG (7 при FDR 5). %, Дополнительный файл 3: Таблица D).Эти гены в основном участвуют в защите и иммунном ответе, но также связаны с усвоением и перевариванием витаминов и метаболизмом арахидоновой кислоты, ключевой жирной кислоты.
Тепловые карты и сети совместного выражения. Тепловые карты корреляций экспрессии генов × генов при направленном отборе (A) и диверсифицирующем отборе (D) соответственно. Каждая строка и столбец представляют собой один и тот же набор генов, аннотированный одной и той же кластерной дендрограммой расстояния экспрессии генов. Кроме того, каждая строка и столбец имеют цветовую кодировку для связанного с ними модуля коэкспрессии генов. На самом графике тепловой карты красный цвет указывает на более похожее совместное выражение, а синий — на большее несходство. Также представлены сети коэкспрессии генов для генов направленного отбора (B) и диверсифицирующего отбора (C) . Узлы взаимодействия были созданы только для генов, которые демонстрируют значительную коэкспрессию при FDR 1%. Черные узлы — это гены, по крайней мере, с 32 значительными взаимодействиями.Красные узлы — это гены, имеющие как минимум семь значимых взаимодействий. Синие узлы — это гены, по крайней мере, с двумя значительными взаимодействиями. Зеленые точки — это гены без существенных взаимодействий при FDR 1%.
Кроме того, каждая строка и столбец имеют цветовую кодировку для связанного с ними модуля коэкспрессии генов. На самом графике тепловой карты красный цвет указывает на более похожее совместное выражение, а синий — на большее несходство. Также представлены сети коэкспрессии генов для генов направленного отбора (B) и диверсифицирующего отбора (C) . Узлы взаимодействия были созданы только для генов, которые демонстрируют значительную коэкспрессию при FDR 1%. Черные узлы — это гены, по крайней мере, с 32 значительными взаимодействиями.Красные узлы — это гены, имеющие как минимум семь значимых взаимодействий. Синие узлы — это гены, по крайней мере, с двумя значительными взаимодействиями. Зеленые точки — это гены без существенных взаимодействий при FDR 1%.
Чтобы оценить, является ли наблюдаемое здесь обогащение продуктом уникальной экспрессии в конкретной популяции или вариации во всех группах, мы разделили все направленные гены по их профилям экспрессии, используя кластеризацию k-средних. При разделении данных профиля экспрессии на две группы (k = 2) мы наблюдаем два противоположных профиля, где экспрессия самая низкая у африканцев, самая высокая у выходцев из Южной и Восточной Азии и промежуточная у европейцев (кластер 1) или самая высокая у африканцев, самая низкая у жителей Южной и выходцы из Восточной Азии, и промежуточное положение у европейцев (кластер 2) (рис. 7, ряд K2).Тесты обогащения для этих двух кластеров показывают, что только кластер 1 демонстрирует какое-либо обогащение, при этом онтология и обогащение путей соответствуют наблюдаемым выше. Это наблюдение согласуется с гипотезой об адаптивных реакциях неафриканских популяций во время миграций из Африки. Однако, когда данные разбиты на большее количество кластеров (k = 6), для тех кластеров, которые подчеркивают различия экспрессии между африканцами и неафриканцами, не происходит обогащения онтологии или пути (рисунок 7, ряд K6, кластеры 4 и 5).Обратите внимание, что мы выбрали K из 6 для этого конкретного анализа, потому что это первый K, который однозначно отделяет африканцев от неафриканских популяций как в повышающей (кластер 4), так и в отрицательной (кластер 5) манере.
При разделении данных профиля экспрессии на две группы (k = 2) мы наблюдаем два противоположных профиля, где экспрессия самая низкая у африканцев, самая высокая у выходцев из Южной и Восточной Азии и промежуточная у европейцев (кластер 1) или самая высокая у африканцев, самая низкая у жителей Южной и выходцы из Восточной Азии, и промежуточное положение у европейцев (кластер 2) (рис. 7, ряд K2).Тесты обогащения для этих двух кластеров показывают, что только кластер 1 демонстрирует какое-либо обогащение, при этом онтология и обогащение путей соответствуют наблюдаемым выше. Это наблюдение согласуется с гипотезой об адаптивных реакциях неафриканских популяций во время миграций из Африки. Однако, когда данные разбиты на большее количество кластеров (k = 6), для тех кластеров, которые подчеркивают различия экспрессии между африканцами и неафриканцами, не происходит обогащения онтологии или пути (рисунок 7, ряд K6, кластеры 4 и 5).Обратите внимание, что мы выбрали K из 6 для этого конкретного анализа, потому что это первый K, который однозначно отделяет африканцев от неафриканских популяций как в повышающей (кластер 4), так и в отрицательной (кластер 5) манере. Результаты от K2 до K8 можно найти в дополнительном файле 1: рисунок S7. Интересно, что именно кластер 1 (рис. 7, ряд K6) с повышенной экспрессией у выходцев из Южной Азии по сравнению с другими группами содержит весь сигнал обогащения. Эти 111 генов обогащены при FDR 20% в 19 путях KEGG (8 при FDR 5%) и 320 категориях GO (136 при FDR 5%).Опять же, они в основном участвуют в иммунном ответе и метаболизме, что согласуется с наблюдениями выше (дополнительный файл 3: таблица E).
Результаты от K2 до K8 можно найти в дополнительном файле 1: рисунок S7. Интересно, что именно кластер 1 (рис. 7, ряд K6) с повышенной экспрессией у выходцев из Южной Азии по сравнению с другими группами содержит весь сигнал обогащения. Эти 111 генов обогащены при FDR 20% в 19 путях KEGG (8 при FDR 5%) и 320 категориях GO (136 при FDR 5%).Опять же, они в основном участвуют в иммунном ответе и метаболизме, что согласуется с наблюдениями выше (дополнительный файл 3: таблица E).
Уровни экспрессии генов, соответствующие направленному отбору. Каждая точка представляет индивидуума, расположенного на расстоянии по оси абсцисс, со средней нормализованной экспрессией гена по оси ординат. Результаты кластерного анализа проиллюстрированы для двух кластеров (К2) и для шести кластеров (К6). Индивидуумы имеют цветовую кодировку по отношению к связанной с ними группе.
При диверсификации генов были идентифицированы три модуля коэкспрессии (рис. 6D) и наблюдались две высокоинтегрированные сети вместе с двумя меньшими сетями (рис. 6C), соответствующие модулям коэкспрессии. Каждый модуль был обогащен многочисленными терминами онтологии GO (дополнительный файл 3: таблица F) и путями KEGG (дополнительный файл 3: таблица G) как с уникальными, так и с перекрывающимися функциями. Модуль 1 (рис. 6D, голубой) обогащен 546 онтологическими терминами GO и 22 путями KEGG при FDR 20% (222 GO и 8 KEGG при FDR 5%) и задействован во многих областях биологии, включая рост, развитие, передачу сигналов, обмен веществ и болезни.Модуль 2 (рис. 6D, синий) обогащен 131 онтологическим термином GO и тремя путями KEGG при FDR 20% (35 GO и 2 KEGG при FDR 5%) и участвует в связывании и взаимодействии с рецептором, в частности, взаимодействие цитокинов с рецепторами цитокинов. и нейроактивное взаимодействие лиганд-рецептор. Модуль 3 (рис. 6D, темно-красный) обогащен 378 онтологическими терминами GO и 12 путями KEGG при FDR 20% (132 GO и 9 KEGG при FDR 5%) и связан с заболеванием и сигнальными путями.
6D) и наблюдались две высокоинтегрированные сети вместе с двумя меньшими сетями (рис. 6C), соответствующие модулям коэкспрессии. Каждый модуль был обогащен многочисленными терминами онтологии GO (дополнительный файл 3: таблица F) и путями KEGG (дополнительный файл 3: таблица G) как с уникальными, так и с перекрывающимися функциями. Модуль 1 (рис. 6D, голубой) обогащен 546 онтологическими терминами GO и 22 путями KEGG при FDR 20% (222 GO и 8 KEGG при FDR 5%) и задействован во многих областях биологии, включая рост, развитие, передачу сигналов, обмен веществ и болезни.Модуль 2 (рис. 6D, синий) обогащен 131 онтологическим термином GO и тремя путями KEGG при FDR 20% (35 GO и 2 KEGG при FDR 5%) и участвует в связывании и взаимодействии с рецептором, в частности, взаимодействие цитокинов с рецепторами цитокинов. и нейроактивное взаимодействие лиганд-рецептор. Модуль 3 (рис. 6D, темно-красный) обогащен 378 онтологическими терминами GO и 12 путями KEGG при FDR 20% (132 GO и 9 KEGG при FDR 5%) и связан с заболеванием и сигнальными путями. Объединение всех диверсифицирующих генов выявляет онтологическое и функциональное обогащение, согласующееся с приведенными выше данными (таблица 4, дополнительный файл 3: таблица H).
Объединение всех диверсифицирующих генов выявляет онтологическое и функциональное обогащение, согласующееся с приведенными выше данными (таблица 4, дополнительный файл 3: таблица H).
Стабилизирующие гены сформировали четыре модуля коэкспрессии, которые как единое целое (Дополнительный файл 3: Таблица I) связаны с 1245 терминами онтологии GO и 51 путями KEGG при FDR 20% (898 GO и 39 KEGG при FDR 20%). 5%) и участвуют в основных, преимущественно внутриклеточных, процессах (таблица 4). К ним относятся ассоциация со сплайсомой, рибосомами, транспортом РНК и процессингом белка.Но они также связаны с неврологическими заболеваниями, такими как болезнь Хантингтона, Паркинсона и болезнь Альцгеймера. Наконец, есть также ассоциации с бактериальной инфекцией, гепатитом С, передачей сигналов Т-клеток и путями развития рака.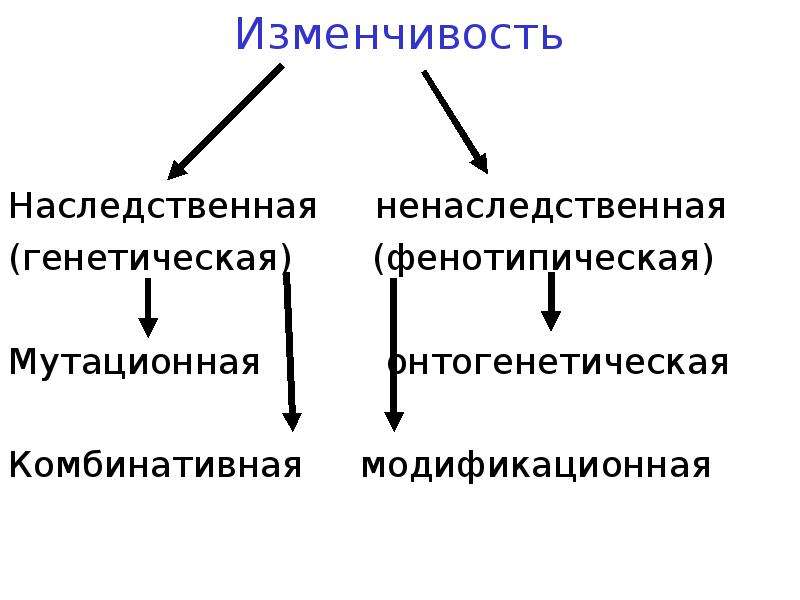 По отдельности каждый модуль имеет уникальный функциональный состав, но несколько ключевых путей, которые включают основные внутриклеточные функции и ассоциации с неврологическими заболеваниями, перекрываются в разной степени (дополнительный файл 3: таблица J и K).
По отдельности каждый модуль имеет уникальный функциональный состав, но несколько ключевых путей, которые включают основные внутриклеточные функции и ассоциации с неврологическими заболеваниями, перекрываются в разной степени (дополнительный файл 3: таблица J и K).
Влияние биологических признаков на экспрессию генов
Наряду с родословной популяции у каждого человека также собирали несколько антропометрических и диетических признаков для оценки их связи с вариациями экспрессии.Начиная с использовавшейся ранее модели экспрессии генов, которая включала технические (количество картированных прочтений и качество РНК) и популяционные факторы (групповые и индивидуальные), были добавлены восемь дополнительных признаков: пол ребенка, вес ребенка, длина ребенок, способ родов (кесарево сечение или вагинальные), возраст матери, индекс массы тела матери, употребляет ли мать алкоголь (вне беременности) и является ли мать вегетарианкой (детали модели см. в разделе «Методы»). Обратите внимание, что каждая новая моделируемая черта является мерой межиндивидуальной изменчивости. Значимость каждого фактора определяли с помощью F-критерия (FDR 5%), используя среднеквадратические оценки каждого фактора по остаточной величине (внутрииндивидуальная вариация).
Значимость каждого фактора определяли с помощью F-критерия (FDR 5%), используя среднеквадратические оценки каждого фактора по остаточной величине (внутрииндивидуальная вариация).
В среднем каждый фактор объясняет примерно 2% вариации данных, при этом большая часть дисперсии приходится на внутрииндивидуальные (32%) и межиндивидуальные (41%) вариации; среди групповых вариаций объясняется 6,3% (рис. 8). Как и ожидалось, подавляющее большинство вариаций, объясняемых каждой из новых объясняющих переменных, ранее объяснялось вариациями среди людей, таким образом, снижение оценки Nit с 0.59 (Nit, рис. 2C) до 0,41 (рис. 8). Все факторы были обогащены не менее чем 59 терминами онтологии GO (дополнительный файл 5: таблица A) при FDR 5%, и все факторы, кроме трех (RIN, пол и длина), были обогащены по крайней мере одним путем KEGG при FDR. 5% (Дополнительный файл 5: Таблица B). Важно отметить, что значимость всех факторов зависела от внутригрупповой и индивидуальной вариации (Nit) и средней экспрессии генов (рис. S7 в дополнительном файле 1). Таким образом, если ген ранее не демонстрировал значительных вариаций среди людей в нашей простой модели генной экспрессии, то он не проявлял значительных вариаций среди любого из восьми дополнительных факторов в нашей полной модели.Таким образом, все термины онтологии GO и пути KEGG, наблюдаемые для каждого из новых факторов, являются просто подмножеством тех, которые ранее были связаны с вариациями среди людей, которое было обогащено 104 путями KEGG и 2720 терминами онтологии GO при FDR 20% ( 65 KEGG, 1729 GO при FDR 5%). С технической стороны, гены, которые коррелировали с количеством картированных прочтений, в подавляющем большинстве случаев были высоко экспрессированы и связаны с такими путями, как рибосомы (KEGG 03010; скорректировано P = 4.75д-23). Известно, что такие технические артефакты являются проблемой этой технологии, и именно поэтому количество картированных прочтений и значения качества РНК (RIN) были включены в качестве ведущих объясняющих переменных во все модели экспрессии генов [43].
S7 в дополнительном файле 1). Таким образом, если ген ранее не демонстрировал значительных вариаций среди людей в нашей простой модели генной экспрессии, то он не проявлял значительных вариаций среди любого из восьми дополнительных факторов в нашей полной модели.Таким образом, все термины онтологии GO и пути KEGG, наблюдаемые для каждого из новых факторов, являются просто подмножеством тех, которые ранее были связаны с вариациями среди людей, которое было обогащено 104 путями KEGG и 2720 терминами онтологии GO при FDR 20% ( 65 KEGG, 1729 GO при FDR 5%). С технической стороны, гены, которые коррелировали с количеством картированных прочтений, в подавляющем большинстве случаев были высоко экспрессированы и связаны с такими путями, как рибосомы (KEGG 03010; скорректировано P = 4.75д-23). Известно, что такие технические артефакты являются проблемой этой технологии, и именно поэтому количество картированных прочтений и значения качества РНК (RIN) были включены в качестве ведущих объясняющих переменных во все модели экспрессии генов [43]. См. Дополнительный файл 5 для всех данных обогащения GO и KEGG для каждого признака.
См. Дополнительный файл 5 для всех данных обогащения GO и KEGG для каждого признака.
График линейки распределения. Каждый ген соответствовал единственной модели, учитывающей 13 объясняющих переменных, и доля изменчивости, объясняемая каждой переменной, оценивалась с использованием метода суммы квадратов.
Одно поразительное наблюдение при подборе модели признаков заключалось в том, что масса тела новорожденного была связана с тремя путями развития рака и ветвью гемопоэтических клеток. Это наблюдение согласуется с сообщениями о том, что вес новорожденного при рождении связан с повышенным риском детской лейкемии [44,45]. Гены, связанные с этим эффектом, подавляются по мере увеличения веса при рождении или усиливаются? Чтобы оценить этот конкретный пример и все другие связанные с ним обогащения признаков, мы разделили корреляции между экспрессией генов и признаками по направлению их действия, а затем повторно оценили ассоциации путей (рис. 9, дополнительный файл 5: таблица C).Результаты указывают на большие скоординированные изменения экспрессии каждого фактора. Например, по мере увеличения массы тела новорожденного снижается экспрессия генов, связанных с линией гемопоэтических клеток, путями развития рака, секрецией желчи, дилатационной кардиомиопатией и сокращением гладкой мускулатуры сосудов, но увеличивается экспрессия генов, связанных с процессингом белков в эндоплазматическом ретикулуме. Кроме того, у людей, которые обычно употребляют алкоголь, снижена экспрессия в таких путях, как гликолиз и переваривание жиров.Плаценты от девочек имеют повышенную экспрессию в переваривании белков, взаимодействии ECM-рецепторов, амебиазе и фокальной адгезии. Плаценты от кесарева сечения демонстрируют пониженную экспрессию гликолиза, процессинга белков в эндоплазматическом ретикулуме и процессинга антигенов. В качестве последнего примера: по мере увеличения индекса массы тела матери происходит коррелированное увеличение экспрессии генов, участвующих в инфицировании золотистым стафилококком, каскадах комплемента и коагуляции, а также в путях системной красной волчанки.
9, дополнительный файл 5: таблица C).Результаты указывают на большие скоординированные изменения экспрессии каждого фактора. Например, по мере увеличения массы тела новорожденного снижается экспрессия генов, связанных с линией гемопоэтических клеток, путями развития рака, секрецией желчи, дилатационной кардиомиопатией и сокращением гладкой мускулатуры сосудов, но увеличивается экспрессия генов, связанных с процессингом белков в эндоплазматическом ретикулуме. Кроме того, у людей, которые обычно употребляют алкоголь, снижена экспрессия в таких путях, как гликолиз и переваривание жиров.Плаценты от девочек имеют повышенную экспрессию в переваривании белков, взаимодействии ECM-рецепторов, амебиазе и фокальной адгезии. Плаценты от кесарева сечения демонстрируют пониженную экспрессию гликолиза, процессинга белков в эндоплазматическом ретикулуме и процессинга антигенов. В качестве последнего примера: по мере увеличения индекса массы тела матери происходит коррелированное увеличение экспрессии генов, участвующих в инфицировании золотистым стафилококком, каскадах комплемента и коагуляции, а также в путях системной красной волчанки. Эти данные, представленные на Фигуре 9, иллюстрируют коррелированный эффект, который изменения экспрессии генов могут оказывать на определенные функциональные пути, а также на физиологию органа или индивидуума.
Эти данные, представленные на Фигуре 9, иллюстрируют коррелированный эффект, который изменения экспрессии генов могут оказывать на определенные функциональные пути, а также на физиологию органа или индивидуума.
Тепловая карта обогащения. Тепловая карта скорректированных Бенджамини-Хохбергом p-значений для связи между каждой независимой переменной (ось X) и категориями пути KEGG (ось Y). Чтобы быть включенным в тепловую карту, путь KEGG должен быть связан по крайней мере с одной объясняющей переменной при FDR 1%.Кроме того, каждая объясняющая переменная была разделена по направлению ее связи с экспрессией гена. Например, переменная «Все овощи. Гены» аннотирует все гены, которые продемонстрировали значительный эффект вегетарианской диеты, в то время как переменная «Увеличение опыта. in Veg.» аннотирует те гены, связанные с вегетарианской диетой, профиль экспрессии которых увеличился по сравнению с невегетарианцами. Точно так же «Поз. Age Genes» аннотирует все гены, которые значительно коррелируют с возрастом матери в положительном ключе.
Точно так же «Поз. Age Genes» аннотирует все гены, которые значительно коррелируют с возрастом матери в положительном ключе.
Инверсия генетической изменчивости в зависимости от пола для приспособленности
Abstract
Поддержание генетической изменчивости приспособленности представляет собой одну из самых давних загадок эволюционной биологии. Половой антагонистический (SA) отбор может в значительной степени способствовать поддержанию генетической изменчивости приспособленности за счет сохранения альтернативных аллелей с противоположными эффектами приспособленности у обоих полов. Это особенно вероятно, если такие локусы SA демонстрируют реверсию доминирования, специфичную для пола (SSDR), где аллель, которая приносит пользу данному полу, также является доминантной для этого пола, что будет генерировать уравновешивающий отбор и поддерживать стабильные полиморфизмы SA для приспособленности.Однако прямые эмпирические тесты SSDR на пригодность в настоящее время отсутствуют. Здесь мы провели полное диаллельное скрещивание среди изогенных штаммов, полученных из естественной популяции семенного жука Callosobruchus maculatus , который, как известно, проявляет генетическую вариабельность SA в приспособленности. Мы измерили специфический для пола репродуктивный успех в течение всей жизни (т.е. приспособленность) в >500 комбинациях F 1 для разных полов и генотипов и обнаружили, что сегрегация генетической изменчивости в приспособленности демонстрирует выраженный вклад дисперсии доминирования и дисперсии доминирования в зависимости от пола.Более тщательное изучение природы дисперсии доминирования показало, что фиксированная аллельная вариация, захваченная в каждом штамме, имеет тенденцию быть доминантной для одного пола, но рецессивной для другого, выявляя полногеномный SSDR для полиморфизмов SA, лежащих в основе приспособленности. Наши результаты показывают, что балансирующий отбор SA может играть недооцененную роль в поддержании изменчивости приспособленности в естественных популяциях.
Здесь мы провели полное диаллельное скрещивание среди изогенных штаммов, полученных из естественной популяции семенного жука Callosobruchus maculatus , который, как известно, проявляет генетическую вариабельность SA в приспособленности. Мы измерили специфический для пола репродуктивный успех в течение всей жизни (т.е. приспособленность) в >500 комбинациях F 1 для разных полов и генотипов и обнаружили, что сегрегация генетической изменчивости в приспособленности демонстрирует выраженный вклад дисперсии доминирования и дисперсии доминирования в зависимости от пола.Более тщательное изучение природы дисперсии доминирования показало, что фиксированная аллельная вариация, захваченная в каждом штамме, имеет тенденцию быть доминантной для одного пола, но рецессивной для другого, выявляя полногеномный SSDR для полиморфизмов SA, лежащих в основе приспособленности. Наши результаты показывают, что балансирующий отбор SA может играть недооцененную роль в поддержании изменчивости приспособленности в естественных популяциях.
Резюме автора
Эволюция требует генетической изменчивости, но отбор будет стремиться зафиксировать те аллели, которые обеспечивают наибольшую приспособленность, истощая генетическую изменчивость, на которую он воздействует.Половая антагонистическая (SA) генетическая изменчивость, при которой альтернативные аллели оказывают противоположное влияние на приспособленность полов, может привести к уравновешивающему отбору, который поддерживает генетическую изменчивость для приспособленности, если аллели, приносящие пользу данному полу, также доминируют у этого пола. Здесь мы показываем, что генетическая вариация SA, лежащая в основе приспособленности в хорошо известной популяции семенных жуков, демонстрирует эти полезные изменения доминирования, предполагая, что отбор SA может обычно поддерживать наследуемую генетическую изменчивость для приспособленности во всем геноме.
Образец цитирования: Grieshop K, Arnqvist G (2018) Изменение генетической изменчивости в зависимости от пола для пригодности. ПЛОС Биол 16(12):
e2006810.
https://doi.org/10.1371/journal.pbio.2006810
ПЛОС Биол 16(12):
e2006810.
https://doi.org/10.1371/journal.pbio.2006810
Академический редактор: Ник Бартон, Австрийский институт науки и технологий, Австрия
Получено: 29 мая 2018 г.; Принято: 27 ноября 2018 г.; Опубликовано: 11 декабря 2018 г.
Copyright: © 2018 Grieshop, Arnqvist.Это статья с открытым доступом, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Доступность данных: Все соответствующие данные находятся в документе и в его файлах вспомогательной информации.
Финансирование: Европейский исследовательский совет (номер гранта GENCON AdG-294333). к ГА. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи. Шведский исследовательский совет (номер гранта 621-2010-5266). к ГА. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи. Шведский исследовательский совет (номер гранта 621-2014-4523). к ГА. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи. Stiftelsenför Zoologisk Forskning (номер гранта). к КГ. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Шведский исследовательский совет (номер гранта 621-2010-5266). к ГА. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи. Шведский исследовательский совет (номер гранта 621-2014-4523). к ГА. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи. Stiftelsenför Zoologisk Forskning (номер гранта). к КГ. Спонсор не участвовал в разработке дизайна исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Конкурирующие интересы: Авторы заявили об отсутствии конкурирующих интересов.
Сокращения: РЭМЛ, ограниченная максимальная вероятность; ЮАР, сексуально антагонистичны; СК, сексуально совместимы; СДР, Отмена доминирования по половому признаку
Введение
Одной из самых давних задач для биологов-эволюционистов было объяснение сохранения генетической изменчивости в приспособленности [1–7]. Отбор должен разрушать генетическую изменчивость, поскольку он устраняет вредные аллели и фиксирует полезные. Тем не менее, природные популяции обладают обильной наследственной изменчивостью в отношении приспособленности и признаков жизненного цикла [8–9]. Двумя общими гипотезами для объяснения этого являются баланс мутаций и отбора и балансирующий отбор [2–4]. В первом случае многие полиморфизмы в геноме поддерживаются на низких частотах аллелей из-за постоянного притока вредных мутаций [10–14], однако этот процесс не может в одиночку объяснить степень и характер генетической изменчивости, наблюдаемой в природе [4]. –7] (поясняется ниже).Таким образом, некоторая форма уравновешивающего отбора, включая сценарии, в которых альтернативные аллели обеспечивают преимущества приспособленности в различных контекстах (например, в условиях окружающей среды, генотипах, временах года или полов), должна способствовать поддержанию полиморфизмов приспособленности во всем геноме [4–7]. .
Отбор должен разрушать генетическую изменчивость, поскольку он устраняет вредные аллели и фиксирует полезные. Тем не менее, природные популяции обладают обильной наследственной изменчивостью в отношении приспособленности и признаков жизненного цикла [8–9]. Двумя общими гипотезами для объяснения этого являются баланс мутаций и отбора и балансирующий отбор [2–4]. В первом случае многие полиморфизмы в геноме поддерживаются на низких частотах аллелей из-за постоянного притока вредных мутаций [10–14], однако этот процесс не может в одиночку объяснить степень и характер генетической изменчивости, наблюдаемой в природе [4]. –7] (поясняется ниже).Таким образом, некоторая форма уравновешивающего отбора, включая сценарии, в которых альтернативные аллели обеспечивают преимущества приспособленности в различных контекстах (например, в условиях окружающей среды, генотипах, временах года или полов), должна способствовать поддержанию полиморфизмов приспособленности во всем геноме [4–7]. .
Половой антагонистический (SA) отбор может привести к тому, что альтернативные аллели будут иметь противоположные эффекты приспособленности у самцов и самок [15–18] и потенциально могут стать наиболее распространенным источником уравновешивающего отбора среди эукариот. Пол — почти повсеместная черта эукариотической жизни [19], генетическая изменчивость SA — неизбежный результат того, что два пола имеют один и тот же геном, но разные оптимумы приспособленности [20–22], а антагонистические формы уравновешивающего отбора должны генерировать более стабильные (менее -транзиентные) полиморфизмы на приспособленность, чем неантагонистические формы балансирующего отбора [23]. Кроме того, хорошо адаптированные популяции должны демонстрировать переизбыток генетической изменчивости SA в отношении приспособленности по сравнению с генетической изменчивостью, согласованной по половому признаку (SC) (т.д., то, что влияет на пол одинаково; см. рис. 1), потому что (1) очищающий отбор должен относительно эффективно удалять генетические вариации СК [24] и (2) скорость, с которой могут разрешаться полиморфизмы СА, должна быть низкой по сравнению со скоростью, с которой возникают новые мутации СК [15, 17–18,25–29]. Растущее количество данных о постоянной генетической изменчивости SA в естественных и лабораторных популяциях в значительной степени подтверждает эти прогнозы (например, [26,30–40]).
Пол — почти повсеместная черта эукариотической жизни [19], генетическая изменчивость SA — неизбежный результат того, что два пола имеют один и тот же геном, но разные оптимумы приспособленности [20–22], а антагонистические формы уравновешивающего отбора должны генерировать более стабильные (менее -транзиентные) полиморфизмы на приспособленность, чем неантагонистические формы балансирующего отбора [23]. Кроме того, хорошо адаптированные популяции должны демонстрировать переизбыток генетической изменчивости SA в отношении приспособленности по сравнению с генетической изменчивостью, согласованной по половому признаку (SC) (т.д., то, что влияет на пол одинаково; см. рис. 1), потому что (1) очищающий отбор должен относительно эффективно удалять генетические вариации СК [24] и (2) скорость, с которой могут разрешаться полиморфизмы СА, должна быть низкой по сравнению со скоростью, с которой возникают новые мутации СК [15, 17–18,25–29]. Растущее количество данных о постоянной генетической изменчивости SA в естественных и лабораторных популяциях в значительной степени подтверждает эти прогнозы (например, [26,30–40]).
Рис. 1. Геометрическое определение терминов и понятий.
Для данного класса наследования Q (например, аддитивность, доминирование, эпистаз и т. д.; см. ниже) BLUP для каждой линии наследования мужского ( q M ) и женского ( q F ) Компоненты дисперсии из моделей, которые были подобраны отдельно для мужских и женских наборов данных, представляют собой оси вариации, которые можно изобразить в виде двумерной зависимости (сплошные линии). Эти системы координат можно повернуть на 45 ° (см. Материалы и методы), чтобы получить измерения SA ( q SA ) и SC ( q SC ) (пунктирные линии) для каждого класса наследования.BLUP, лучший линейный несмещенный прогноз; SA, сексуально антагонистический; SC, сексуально конкордантный.
https://doi.org/10.1371/journal.pbio.2006810.g001
Способность СА-отбора генерировать уравновешивающий отбор, который приводит к стабильным полиморфизмам приспособленности, резко возрастает с реверсией доминирования по признаку пола (SSDR) [41–42]. ], в котором аллели, которые способствуют приспособленности данного пола, также доминируют в этом поле, создавая чистое преимущество гетерозигот в популяции.Такие полезные изменения доминирования в более общем случае антагонистической плейотропии [43] были встречены с ранним скептицизмом [44-46], но более поздняя теория меняет эту точку зрения. Ожидается, что средний локус, лежащий в основе двух полигенных гомологичных фенотипов с антагонистическими эффектами приспособленности (например, мужской и женской приспособленности), будет демонстрировать, по крайней мере, частичную инверсию доминирования приспособленности при неограничительном предположении, что функции приспособленности вогнуты вблизи их перекрытия [47]. ].Кроме того, генетическая вариация SA должна, в свою очередь, выбирать локусы-модификаторы, которые позволяют гетерозиготным мужчинам и женщинам демонстрировать благоприятные отношения доминирования между аллелями SA [48], что делает SSDR генетической изменчивости SA для приспособленности прогнозируемым результатом адаптивной эволюции.
], в котором аллели, которые способствуют приспособленности данного пола, также доминируют в этом поле, создавая чистое преимущество гетерозигот в популяции.Такие полезные изменения доминирования в более общем случае антагонистической плейотропии [43] были встречены с ранним скептицизмом [44-46], но более поздняя теория меняет эту точку зрения. Ожидается, что средний локус, лежащий в основе двух полигенных гомологичных фенотипов с антагонистическими эффектами приспособленности (например, мужской и женской приспособленности), будет демонстрировать, по крайней мере, частичную инверсию доминирования приспособленности при неограничительном предположении, что функции приспособленности вогнуты вблизи их перекрытия [47]. ].Кроме того, генетическая вариация SA должна, в свою очередь, выбирать локусы-модификаторы, которые позволяют гетерозиготным мужчинам и женщинам демонстрировать благоприятные отношения доминирования между аллелями SA [48], что делает SSDR генетической изменчивости SA для приспособленности прогнозируемым результатом адаптивной эволюции.
Способность SSDR способствовать поддержанию полиморфизмов SA для приспособленности известна уже несколько десятилетий [41], однако в настоящее время нет прямых эмпирических тестов SSDR на пригодность. Барсон и его коллеги [39] недавно продемонстрировали SSDR для одного локуса основного эффекта, влияющего на возраст половозрелости у лосося.Приспособленность, однако, является очень полигенным признаком, и исследование свойств многих локусов обычно требует количественного генетического подхода [49-50].
Полное диаллельное скрещивание [50] является основным методом количественного генетического разведения, способным разделить фенотипическую изменчивость на вариации, связанные с аддитивными генетическими эффектами, родительскими эффектами, доминированием и эпистазом. Это позволяет проверить несколько гипотез о сохранении генетической изменчивости приспособленности. Поскольку многие слабо вредные аллели, поддерживаемые балансом мутаций и отбора, обязательно должны демонстрировать частичное доминирование [4, 7, 10–14, 51–52], они должны генерировать выраженную аддитивную генетическую изменчивость по отношению к дисперсии доминирования, если они вносят основной вклад в приспособленность. дисперсия.Напротив, уравновешивающий отбор, действующий для поддержания относительно меньшего количества полиморфизмов с большей величиной эффекта, который может включать коэффициенты доминирования вплоть до полного доминирования включительно, приведет к выраженной дисперсии доминирования по сравнению с аддитивной генетической изменчивостью, если она преобладает [4,7–8,50]. . Что касается различия между многими формами выбора балансировки, данные dialllel предлагают возможность отличить выбор балансировки SA от других форм выбора балансировки путем тестирования его отличительной черты SSDR (см. выше).С этой целью разделение по дисперсии не является достаточным тестом. Скорее, полные диаллельные данные дают возможность количественно определить относительное количество фиксированных рецессивных аллельных вариаций среди набора инбредных штаммов, а если данные зависят от пола, то это позволяет сделать это независимо для самцов и самок. Положительная корреляция между штаммами относительно их относительного количества рецессивных аллельных вариаций при измерении у самцов по сравнению с самками будет демонстрировать, что штаммы имеют тенденцию демонстрировать относительно высокое или низкое количество фиксированных рецессивных аллелей (независимо от того, в каком поле они экспрессируются), тогда как отрицательная Корреляция показала бы, что штаммы склонны фиксировать аллельную изменчивость, которая является рецессивной у одного пола, но доминантной у другого (т.
дисперсия.Напротив, уравновешивающий отбор, действующий для поддержания относительно меньшего количества полиморфизмов с большей величиной эффекта, который может включать коэффициенты доминирования вплоть до полного доминирования включительно, приведет к выраженной дисперсии доминирования по сравнению с аддитивной генетической изменчивостью, если она преобладает [4,7–8,50]. . Что касается различия между многими формами выбора балансировки, данные dialllel предлагают возможность отличить выбор балансировки SA от других форм выбора балансировки путем тестирования его отличительной черты SSDR (см. выше).С этой целью разделение по дисперсии не является достаточным тестом. Скорее, полные диаллельные данные дают возможность количественно определить относительное количество фиксированных рецессивных аллельных вариаций среди набора инбредных штаммов, а если данные зависят от пола, то это позволяет сделать это независимо для самцов и самок. Положительная корреляция между штаммами относительно их относительного количества рецессивных аллельных вариаций при измерении у самцов по сравнению с самками будет демонстрировать, что штаммы имеют тенденцию демонстрировать относительно высокое или низкое количество фиксированных рецессивных аллелей (независимо от того, в каком поле они экспрессируются), тогда как отрицательная Корреляция показала бы, что штаммы склонны фиксировать аллельную изменчивость, которая является рецессивной у одного пола, но доминантной у другого (т. д., СДР). Вполне возможно, что SSDR такого рода могут развиваться в контексте аллельных вариантов SC, которые просто демонстрируют половые различия в относительной силе отбора, действующего на каждый из них. Таким образом, наиболее консервативным и явным тестом SSDR на генетическую вариацию SA, лежащую в основе приспособленности, будет анализ этой корреляции после статистического удаления аддитивных генетических эффектов SC из данных.
д., СДР). Вполне возможно, что SSDR такого рода могут развиваться в контексте аллельных вариантов SC, которые просто демонстрируют половые различия в относительной силе отбора, действующего на каждый из них. Таким образом, наиболее консервативным и явным тестом SSDR на генетическую вариацию SA, лежащую в основе приспособленности, будет анализ этой корреляции после статистического удаления аддитивных генетических эффектов SC из данных.
Здесь мы использовали полное диаллельное скрещивание среди 16 изогенных штаммов, чтобы разделить генетическую изменчивость в половом репродуктивном успехе в течение всей жизни (далее «приспособленность») в выловленной в дикой природе популяции семенного жука C . maculatus , который, как известно, демонстрирует выраженную генетическую изменчивость SA в приспособленности [37–38,53]. Этот вид демонстрирует полиандрическую систему спаривания, детерминацию пола X/Y, выраженный половой диморфизм и экспрессию генов, зависящих от пола [33,38,54–56]. В общей сложности было проведено 3 278 индивидуальных анализов приспособленности (1 731 мужчина и 1 547 женщин) по 256 возможным типам скрещивания (далее «семейства»), т. е. 240 ауткроссированных (гетерозиготных) семей и 16 родительских (гомозиготных) семей полного 16 × 16 диаллель.Учитывая, что эти инбредные штаммы происходят из популяции, чья генетическая изменчивость в приспособленности является преимущественно SA [37], настоящие результаты выраженной дисперсии доминирования и дисперсии доминирования по полу по сравнению с аддитивной генетической изменчивостью (рис. 2) позволяют предположить, что дисперсия приспособленности этой популяции составляет в значительной степени основано на относительно небольшом количестве полиморфизмов с большим эффектом при отборе SA, в отличие от многих полиморфизмов с малым эффектом в балансе мутация-селекция [4,7-8,50]. Наши анализы показали, что штаммы демонстрировали значительно отрицательную ранговую корреляцию для их относительного количества фиксированных доминантных аллелей приспособленности при измерении у самцов по сравнению с самками (рис.
В общей сложности было проведено 3 278 индивидуальных анализов приспособленности (1 731 мужчина и 1 547 женщин) по 256 возможным типам скрещивания (далее «семейства»), т. е. 240 ауткроссированных (гетерозиготных) семей и 16 родительских (гомозиготных) семей полного 16 × 16 диаллель.Учитывая, что эти инбредные штаммы происходят из популяции, чья генетическая изменчивость в приспособленности является преимущественно SA [37], настоящие результаты выраженной дисперсии доминирования и дисперсии доминирования по полу по сравнению с аддитивной генетической изменчивостью (рис. 2) позволяют предположить, что дисперсия приспособленности этой популяции составляет в значительной степени основано на относительно небольшом количестве полиморфизмов с большим эффектом при отборе SA, в отличие от многих полиморфизмов с малым эффектом в балансе мутация-селекция [4,7-8,50]. Наши анализы показали, что штаммы демонстрировали значительно отрицательную ранговую корреляцию для их относительного количества фиксированных доминантных аллелей приспособленности при измерении у самцов по сравнению с самками (рис. 3).Таким образом, является ли средний аллель, лежащий в основе приспособленности в этой популяции, доминантным или рецессивным у гетерозиготы, зависит от того, экспрессируется ли он у самца или самки. Как упоминалось выше, это все еще могло иметь место и для некоторых аллельных вариаций SC, но эта связь оставалась сильной, когда аддитивные генетические эффекты SC были предварительно статистически удалены из данных (рис. S1), явно демонстрируя SSDR для генетической вариации SA, лежащей в основе фитнес. Наши результаты согласуются с тем, что SSDR для всего генома поддерживает сбалансированные полиморфизмы SA для приспособленности, что имеет важные последствия для способности отбора SA объяснять изменчивость приспособленности в естественных популяциях.
3).Таким образом, является ли средний аллель, лежащий в основе приспособленности в этой популяции, доминантным или рецессивным у гетерозиготы, зависит от того, экспрессируется ли он у самца или самки. Как упоминалось выше, это все еще могло иметь место и для некоторых аллельных вариаций SC, но эта связь оставалась сильной, когда аддитивные генетические эффекты SC были предварительно статистически удалены из данных (рис. S1), явно демонстрируя SSDR для генетической вариации SA, лежащей в основе фитнес. Наши результаты согласуются с тем, что SSDR для всего генома поддерживает сбалансированные полиморфизмы SA для приспособленности, что имеет важные последствия для способности отбора SA объяснять изменчивость приспособленности в естественных популяциях.
Рис. 2. Графическое представление разбиения по дисперсии.
Компоненты дисперсии ( σ 2 , ± 1 с.э.) для пригодности, оцененной с помощью REML. Данные, лежащие в основе всех рисунков и таблиц, можно найти в S1 Data. асимм., асимметричный; эфф., эффекты; эпи., эпистаз; REML, ограниченная максимальная вероятность; с., стандартная ошибка.
асимм., асимметричный; эфф., эффекты; эпи., эпистаз; REML, ограниченная максимальная вероятность; с., стандартная ошибка.
https://doi.org/10.1371/journal.pbio.2006810.g002
Рис. 3. SSDR аллельной вариации, лежащей в основе приспособленности.
(A) Относительное количество рецессивных аллельных вариаций приспособленности у самцов () и самок () значительно отрицательно коррелировало ( = -0,779 [95% ДИ от -0,92 до -0,46], P = 0,0004) между линиями ( N = 16; единицы отражают нестандартизированную остаточную приспособленность из модели, учитывающей средовую и эпистатическую изменчивость), и (B) то же самое отношение, проиллюстрированное и проанализированное как ранги (т. е. штаммы, ранжированные в порядке их относительного «доминирования» над одним другой; : −0.738, P = 0,0016). Штаммы имели тенденцию к обогащению аллельными вариациями приспособленности, которые были доминантными у их гетерозиготных сыновей, но рецессивными у их гетерозиготных дочерей, и наоборот. SSDR, инверсия доминирования по половому признаку.
SSDR, инверсия доминирования по половому признаку.
https://doi.org/10.1371/journal.pbio.2006810.g003
Результаты
Мы провели полное диаллельное скрещивание среди 16 изогенных штаммов, получив поколение F 1 из 240 возможных ауткроссированных семей и 16 родительских самов.Мы проанализировали репродуктивный успех самцов и самок F 1 в течение всей жизни (т. е. приспособленность) отдельно, чтобы получить специфические для пола показатели приспособленности для каждой ауткроссированной семьи и самоопыленной линии (рис. S2). Фенотипическая изменчивость приспособленности была разделена на вариации, связанные с общими фиксированными эффектами блока репликации ( x ), инбридинга ( b 1 ), пола ( S ) и инбридинга по признаку пола ( S × b 1 ), а также следующие штаммо- и кросс-специфические случайные эффекты (т.е., компоненты дисперсии) с использованием оценки ограниченного максимального правдоподобия (REML): аддитивная генетическая дисперсия ( a ), доминирование ( b 2 ), родительские эффекты ( c ), симметричный эпистаз ( b 3 ), асимметричный эпистаз ( d ) и их половые варианты (т. е. их взаимодействие с S ). Для этого подхода REML мы использовали процедуру FDIALLEL [57] для GenStat (v.18.2.0.18409; [58]), чтобы подогнать пользовательскую версию модели Хеймана [59], которую мы модифицировали для учета эффектов, специфичных для пола (далее « «полнополая» модель).Мы также применили полнополую иерархическую байесовскую модель Ленарчича и его коллег [60] к нашим данным, используя сэмплер «BayesDiallel» MCMC Gibbs [61] для R (v.3.2.1; [62]). Два подхода дали качественно схожие результаты (байесовский подход представлен на рисунках S3–S5, в таблицах S1 и S2 и в тексте S1).
е. их взаимодействие с S ). Для этого подхода REML мы использовали процедуру FDIALLEL [57] для GenStat (v.18.2.0.18409; [58]), чтобы подогнать пользовательскую версию модели Хеймана [59], которую мы модифицировали для учета эффектов, специфичных для пола (далее « «полнополая» модель).Мы также применили полнополую иерархическую байесовскую модель Ленарчича и его коллег [60] к нашим данным, используя сэмплер «BayesDiallel» MCMC Gibbs [61] для R (v.3.2.1; [62]). Два подхода дали качественно схожие результаты (байесовский подход представлен на рисунках S3–S5, в таблицах S1 и S2 и в тексте S1).
Для облегчения интерпретации были также выполнены отдельные мужские и женские модели (на отдельных мужских и женских наборах данных), которые предоставили компоненты дисперсии по половому признаку рис. 1), которые использовались в следующей геометрической интерпретации «половых» и «неполовых» компонентов дисперсии из полнополовой модели (см. ниже) для всех классов наследования (см. Таблицу S3 для отдельных оценок компонентов дисперсии, специфичных для пола).Для заданного класса наследования Q (например, аддитивность, доминирование, эпистаз и т. д.) полнополая модель обеспечивает половые ( S × q ) и неполовые ( q ) компоненты дисперсии. Лучшие линейные несмещенные прогнозы (BLUP) штаммов для данного компонента дисперсии представляют их оценочные значения вдоль этих осей вариации. Все неполовые оси изменчивости ( q ) из полнополой модели сильно коррелировали (т. е. ≈) с производной размерностью SC их соответствующего класса наследования ( q SC ; все коэффициенты корреляции Пирсона были > 0 .98, все значения P <0,0001; см. Материалы и методы и Рис. 1). Таким образом, было обнаружено, что все нераспределенные по полу компоненты дисперсии из модели с полным разделением по полу представляют эффекты SC. Это было верно даже в случаях технически неправильных корреляций, в которых стандартная ошибка q , q M или q F перекрывалась с нулем (например, = 923 P ).
Таблицу S3 для отдельных оценок компонентов дисперсии, специфичных для пола).Для заданного класса наследования Q (например, аддитивность, доминирование, эпистаз и т. д.) полнополая модель обеспечивает половые ( S × q ) и неполовые ( q ) компоненты дисперсии. Лучшие линейные несмещенные прогнозы (BLUP) штаммов для данного компонента дисперсии представляют их оценочные значения вдоль этих осей вариации. Все неполовые оси изменчивости ( q ) из полнополой модели сильно коррелировали (т. е. ≈) с производной размерностью SC их соответствующего класса наследования ( q SC ; все коэффициенты корреляции Пирсона были > 0 .98, все значения P <0,0001; см. Материалы и методы и Рис. 1). Таким образом, было обнаружено, что все нераспределенные по полу компоненты дисперсии из модели с полным разделением по полу представляют эффекты SC. Это было верно даже в случаях технически неправильных корреляций, в которых стандартная ошибка q , q M или q F перекрывалась с нулем (например, = 923 P ).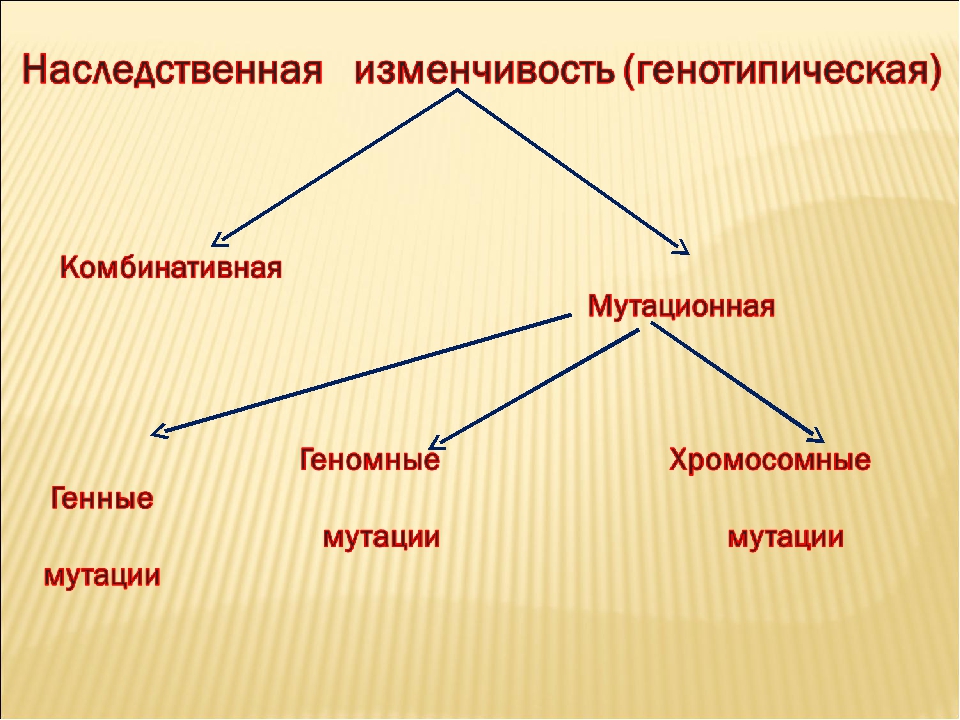 0,0001, несмотря на то, что и равны приблизительно 0; см. ниже).Кроме того, компоненты половой дисперсии для аддитивности ( S × a ), доминирования ( S × b 2 ) и эпистаза ( S × b 9009) представляют10 0 . Эффекты SA: внутренний продукт между этими эффектами без пола (т.е. SC, см. выше) и эффектами с сексом показал, что они описывают приблизительно ортогональные оси вариации (отображая последний SA; см. рис. 1). То есть их BLUP падали по осям вариации, которые находились под угловым смещением примерно 90 °, θ , друг от друга ( θ a , S × a = 91.50°, = 90,00°, = 91,49°). Отрицательные оценки компонента дисперсии для родительских эффектов ( c ) и специфического для пола асимметричного эпистаза ( S × d ) сделали их BLUP недействительными и исключили их классы наследования из этой геометрической интерпретации.
0,0001, несмотря на то, что и равны приблизительно 0; см. ниже).Кроме того, компоненты половой дисперсии для аддитивности ( S × a ), доминирования ( S × b 2 ) и эпистаза ( S × b 9009) представляют10 0 . Эффекты SA: внутренний продукт между этими эффектами без пола (т.е. SC, см. выше) и эффектами с сексом показал, что они описывают приблизительно ортогональные оси вариации (отображая последний SA; см. рис. 1). То есть их BLUP падали по осям вариации, которые находились под угловым смещением примерно 90 °, θ , друг от друга ( θ a , S × a = 91.50°, = 90,00°, = 91,49°). Отрицательные оценки компонента дисперсии для родительских эффектов ( c ) и специфического для пола асимметричного эпистаза ( S × d ) сделали их BLUP недействительными и исключили их классы наследования из этой геометрической интерпретации. Опять же, все неполовые компоненты дисперсии описывают эффекты SC, в то время как аддитивный ( S × a ), доминантный ( S × b 2 ) и эпистатический ( S × 1 b 3 ) компоненты дисперсии (ортогональные своим аналогам SC) описывают эффекты SA (см. рис. 1).Поэтому мы ссылаемся на них соответствующим образом (например, аддитивная генетическая дисперсия SA). Дополнительная информация о терминологии и значении компонентов дисперсии доступна в тексте S1.
Опять же, все неполовые компоненты дисперсии описывают эффекты SC, в то время как аддитивный ( S × a ), доминантный ( S × b 2 ) и эпистатический ( S × 1 b 3 ) компоненты дисперсии (ортогональные своим аналогам SC) описывают эффекты SA (см. рис. 1).Поэтому мы ссылаемся на них соответствующим образом (например, аддитивная генетическая дисперсия SA). Дополнительная информация о терминологии и значении компонентов дисперсии доступна в тексте S1.
Дисперсионное разделение
Не было общих различий между полами ( S ) в средней приспособленности (Таблица 1). Общий эффект инбридинга ( b 1 ) был большим и статистически значимым (таблица 1). Полы различались в этом отношении, о чем свидетельствует значительный половой инбридинг ( S × b 1 ; табл. 1).Отдельные мужские и женские модели показали, что эффект инбридинга был сильнее у мужчин, чем у женщин (таблица S3), что подтверждается другими исследованиями (например, [63]). Обратите внимание, что эти заметные эффекты инбридинга и инбридинга по признаку пола учитывались как общие (фиксированные) эффекты до оценки компонентов дисперсии случайных эффектов и, следовательно, не завышают и не нарушают оценки доминирования ( b 2 ), пола -специфическое доминирование ( S × b 2 ) или любой другой компонент дисперсии.
Обратите внимание, что эти заметные эффекты инбридинга и инбридинга по признаку пола учитывались как общие (фиксированные) эффекты до оценки компонентов дисперсии случайных эффектов и, следовательно, не завышают и не нарушают оценки доминирования ( b 2 ), пола -специфическое доминирование ( S × b 2 ) или любой другой компонент дисперсии.
Таблица 1. Результаты полной модели REML с разделением по полу.
F-статистика с P значения для общих (фиксированных) эффектов и дисперсий ( σ 2 ) с s.e. для штаммовых и кросс-специфических (случайных) эффектов (т. е. компонентов дисперсии).
https://doi.org/10.1371/journal.pbio.2006810.t001
В целом наследование приспособленности характеризовалось выраженным доминированием SC ( b 2 ) и доминированием SA ( S × b 2 , таблица 1 и рис. 2).Также был относительно небольшой, но ненулевой вклад эпистатической дисперсии SC ( b 3 ; таблица 1 и рис. 2). Из них b 2 и S × b 2 соответственно вносили примерно 17-кратный и примерно 8-кратный вклад в генетическую дисперсию приспособленности по сравнению с b 3 (Таблица 100).
2). Из них b 2 и S × b 2 соответственно вносили примерно 17-кратный и примерно 8-кратный вклад в генетическую дисперсию приспособленности по сравнению с b 3 (Таблица 100).
Хотя стандартная ошибка аддитивной генетической дисперсии SA ( S × a ) едва перекрывала ноль, мы отмечаем, что ее оценка была в 10 раз выше, чем у аддитивной генетической дисперсии SC ( a ) (таблица 1 и рис. 2). .Родительские эффекты ( c ), половые родительские эффекты ( S × c ), SA эпистаз ( S × b 3 ), асимметричный эпистаз ( d ) и половой специфичный асимметричный эпистаз ( S × d ) был оценен как близкий к нулю со стандартными ошибками, перекрывающими ноль (таблица 1 и рис. 2).
Отдельные мужские и женские модели показали примерно в 5 раз большую остаточную дисперсию ( ε ) для мужчин, чем для женщин, что усложняет точное количественное сравнение между этими моделями (таблица S3). Однако качественно ненулевая дисперсия была оценена для доминирования ( b 2 ) и эпистаза ( b 3 ) как в мужских, так и в женских моделях, и это были единственные классы наследования с ненулевой дисперсией в обеих моделях (S3 Таблица). В обеих моделях b 2 оценивается примерно в 13 раз больше, чем b 3 (таблица S3).
Однако качественно ненулевая дисперсия была оценена для доминирования ( b 2 ) и эпистаза ( b 3 ) как в мужских, так и в женских моделях, и это были единственные классы наследования с ненулевой дисперсией в обеих моделях (S3 Таблица). В обеих моделях b 2 оценивается примерно в 13 раз больше, чем b 3 (таблица S3).
Оценка SSDR
Полное диаллельное скрещивание изогенных штаммов дает дополнительную ценную информацию о доминантных отношениях между фиксированными аллельными вариантами среди штаммов [50,59].В частности, оценка относительной доли доминантных: рецессивных аллелей среди гомозиготных родительских линий дается набором ковариаций между средними семейными значениями ауткроссированных (гетерозиготных) штаммов и средними значениями инбредных (гомозиготных) родительских «я» линий, к которым они были скрещены (S6 Fig [50,59]). Штаммы, чьи ауткроссированные (гетерозиготные) семейные средства определяются (т. е. ковариацией) инбредными (гомозиготными) средствами штаммов, с которыми они скрещиваются, несут аллели, явно рецессивные по отношению к аллелям других штаммов.Напротив, штаммы, чьи ауткроссированные (гетерозиготные) семейные средства вместо этого независимы (т. Е. Не коррелируют с) инбредных (гомозиготных) средств штаммов, с которыми они скрещиваются, содержат аллели, которые явно доминируют над аллелями других штаммов. После удаления из данных экологических и эпистатических вариаций, σ P , r , ковариация каждого штамма между его ауткроссированными семейными средствами ( r ) и средними значениями инбредных родительских самостей ( P ) штаммы, с которыми их скрещивали [50,59], использовали в качестве оценки относительного количества рецессивных аллелей, зафиксированных внутри каждого штамма (см. Материалы и методы и S6 Fig).Это было сделано для пригодности мужчин () и женщин () отдельно, и эти оценки имели значительную отрицательную корреляцию (Pearson = -0,779 (95% ДИ от -0,92 до -0,46), P = 0,0004; рис.
е. ковариацией) инбредными (гомозиготными) средствами штаммов, с которыми они скрещиваются, несут аллели, явно рецессивные по отношению к аллелям других штаммов.Напротив, штаммы, чьи ауткроссированные (гетерозиготные) семейные средства вместо этого независимы (т. Е. Не коррелируют с) инбредных (гомозиготных) средств штаммов, с которыми они скрещиваются, содержат аллели, которые явно доминируют над аллелями других штаммов. После удаления из данных экологических и эпистатических вариаций, σ P , r , ковариация каждого штамма между его ауткроссированными семейными средствами ( r ) и средними значениями инбредных родительских самостей ( P ) штаммы, с которыми их скрещивали [50,59], использовали в качестве оценки относительного количества рецессивных аллелей, зафиксированных внутри каждого штамма (см. Материалы и методы и S6 Fig).Это было сделано для пригодности мужчин () и женщин () отдельно, и эти оценки имели значительную отрицательную корреляцию (Pearson = -0,779 (95% ДИ от -0,92 до -0,46), P = 0,0004; рис. 3A). Этот эффект не был обусловлен каким-либо конкретным штаммом (штаммами), о чем свидетельствует значительно отрицательная непараметрическая ранговая корреляция, указывающая на относительное «доминирование» штаммов друг над другом (Спирмен: -0,738, P = 0,0016; рис. 3B). Эти тесты оставались сильно значимыми после того, как сначала была статистически удалена аддитивная генетическая дисперсия SC (Pearson’s = -0.665 (95% ДИ от -0,87 до -0,25), P = 0,005; Спирмена: -0,635, P = 0,0098; S1 Fig), явно демонстрирующий SSDR генетической вариации SA для приспособленности.
3A). Этот эффект не был обусловлен каким-либо конкретным штаммом (штаммами), о чем свидетельствует значительно отрицательная непараметрическая ранговая корреляция, указывающая на относительное «доминирование» штаммов друг над другом (Спирмен: -0,738, P = 0,0016; рис. 3B). Эти тесты оставались сильно значимыми после того, как сначала была статистически удалена аддитивная генетическая дисперсия SC (Pearson’s = -0.665 (95% ДИ от -0,87 до -0,25), P = 0,005; Спирмена: -0,635, P = 0,0098; S1 Fig), явно демонстрирующий SSDR генетической вариации SA для приспособленности.
Обсуждение
Прогресс в нашем общем понимании поддержания генетической изменчивости в приспособленности требует выявления широко применимых механизмов. Зная, что баланс мутаций и отбора не может объяснить всего [4–7], многие биологи-эволюционисты задаются вопросом: какие формы балансирующего отбора объясняют оставшуюся генетическую изменчивость? Существует много механизмов уравновешивания отбора с теоретической и эмпирической поддержкой, которые способны поддерживать стабильный полиморфизм для приспособленности при определенных условиях. Тем не менее, отбор SA потенциально является одним из самых распространенных среди эукариот — в ожидании преобладания SSDR — предлагая общее решение этого классического эволюционного вопроса. К сожалению, отбор SA часто не упоминается в обзорах по этой теме (например, [5–6]), что делает его относительно плохо изученным с точки зрения современной геномики [64–65]. Поэтому мы кратко рассмотрим неизбежность генетической изменчивости SA, для которой настоящие данные и другие недавние исследования существенно усиливают аргументы в пользу ее роли в поддержании генетической изменчивости в приспособленности.
Тем не менее, отбор SA потенциально является одним из самых распространенных среди эукариот — в ожидании преобладания SSDR — предлагая общее решение этого классического эволюционного вопроса. К сожалению, отбор SA часто не упоминается в обзорах по этой теме (например, [5–6]), что делает его относительно плохо изученным с точки зрения современной геномики [64–65]. Поэтому мы кратко рассмотрим неизбежность генетической изменчивости SA, для которой настоящие данные и другие недавние исследования существенно усиливают аргументы в пользу ее роли в поддержании генетической изменчивости в приспособленности.
Антагонистические мутации (демонстрирующие любую форму антагонистической плейотропии) медленнее закрепляются, чем неантагонистические мутации, а это означает, что они будут генерировать более слабую геномную сигнатуру отбора, но на самом деле будут вносить больший и более устойчивый вклад в генетическую изменчивость приспособленности [23]. Соответственно, экспериментальная эволюция микроорганизмов часто показывает, что адаптация в данном контексте или окружающей среде происходит за счет снижения приспособленности в других контекстах (обзор в [66]), что указывает на существенную постоянную генетическую изменчивость с антагонистическими плейотропными эффектами. Аналогично, Qian и коллеги [67] продемонстрировали широко распространенную антагонистическую плейотропию в сотнях генов в геноме дрожжей. Ожидается, что распространенность этих генетических компромиссов будет увеличиваться с увеличением сложности организма, такой как специализированные ткани, стадии развития и пол [67-69]. Кроме того, разрешение таких генетических компромиссов развивается медленнее при меньших эффективных размерах популяции и при более длительном времени генерации [67], что опять же подразумевает еще большую вероятность того, что антагонистическая плейотропия будет поддерживать генетическую изменчивость в приспособленности в сложных многоклеточных организмах (т.д., эукариоты). Таким образом, учитывая, что половое размножение является почти повсеместным признаком эукариотической жизни [19], пол, вероятно, представляет собой наиболее последовательный и широко распространенный набор контекстов, в которых может возникнуть антагонистическая плейотропия.
Аналогично, Qian и коллеги [67] продемонстрировали широко распространенную антагонистическую плейотропию в сотнях генов в геноме дрожжей. Ожидается, что распространенность этих генетических компромиссов будет увеличиваться с увеличением сложности организма, такой как специализированные ткани, стадии развития и пол [67-69]. Кроме того, разрешение таких генетических компромиссов развивается медленнее при меньших эффективных размерах популяции и при более длительном времени генерации [67], что опять же подразумевает еще большую вероятность того, что антагонистическая плейотропия будет поддерживать генетическую изменчивость в приспособленности в сложных многоклеточных организмах (т.д., эукариоты). Таким образом, учитывая, что половое размножение является почти повсеместным признаком эукариотической жизни [19], пол, вероятно, представляет собой наиболее последовательный и широко распространенный набор контекстов, в которых может возникнуть антагонистическая плейотропия.
Несмотря на то, что у представителей обоих полов в основном один и тот же геном, они приобретают приспособленность по-разному, а это означает, что их оптимумы приспособленности для ряда признаков жизненного цикла обычно различаются [16–18,22]. Таким образом, точно так же, как геометрическая модель Фишера [1] предсказывает, что подавляющее большинство мутаций будут вредными, редкая мутация с преимуществами приспособленности у одного пола будет иметь тенденцию создавать недостатки приспособленности у другого пола — общий геном, таким образом, делает генетические вариации SA неизбежными [20]. –22].Затем такие антагонистические мутации будут иметь тенденцию к достижению промежуточных равновесных частот аллелей [23], в то время как очищающий отбор будет иметь тенденцию к устранению мутаций, которые генерируют SC генетическую изменчивость, оставляя после себя в основном SA генетическую изменчивость в приспособленности [24] (см. рис. 1). Кроме того, ограничение, которое общий геном накладывает на адаптивную эволюцию, специфичную для пола, подразумевает, что существует лишь ограниченная степень, в которой полиморфизмы SA могут быть разрешены по сравнению со скоростью, с которой возникают новые мутации SA [17–18,25–29]. , что согласуется с растущим объемом эмпирических данных о постоянной генетической изменчивости SA (например,г.
Таким образом, точно так же, как геометрическая модель Фишера [1] предсказывает, что подавляющее большинство мутаций будут вредными, редкая мутация с преимуществами приспособленности у одного пола будет иметь тенденцию создавать недостатки приспособленности у другого пола — общий геном, таким образом, делает генетические вариации SA неизбежными [20]. –22].Затем такие антагонистические мутации будут иметь тенденцию к достижению промежуточных равновесных частот аллелей [23], в то время как очищающий отбор будет иметь тенденцию к устранению мутаций, которые генерируют SC генетическую изменчивость, оставляя после себя в основном SA генетическую изменчивость в приспособленности [24] (см. рис. 1). Кроме того, ограничение, которое общий геном накладывает на адаптивную эволюцию, специфичную для пола, подразумевает, что существует лишь ограниченная степень, в которой полиморфизмы SA могут быть разрешены по сравнению со скоростью, с которой возникают новые мутации SA [17–18,25–29]. , что согласуется с растущим объемом эмпирических данных о постоянной генетической изменчивости SA (например,г. , [26,30–40]). Наконец, даже половые различия в силе отбора альтернативных аллельных вариантов, лежащих в основе SC-форм антагонистической плейотропии между различными компонентами приспособленности (например, выживаемость, плодовитость, фертильность, конкуренция партнеров и т. д.), могут привести к тому, что аллельные компромиссы будут иметь эффекты SA. на общую приспособленность [70], предполагая, что (1) вероятность генетической вариации SA для приспособленности даже выше, чем предполагала предыдущая теория, и (2) исследования, в которых отсутствуют доказательства генетической изменчивости SA для данного компонента приспособленности, не говорят об этом. наличие или отсутствие генетической вариации SA для приспособленности.
, [26,30–40]). Наконец, даже половые различия в силе отбора альтернативных аллельных вариантов, лежащих в основе SC-форм антагонистической плейотропии между различными компонентами приспособленности (например, выживаемость, плодовитость, фертильность, конкуренция партнеров и т. д.), могут привести к тому, что аллельные компромиссы будут иметь эффекты SA. на общую приспособленность [70], предполагая, что (1) вероятность генетической вариации SA для приспособленности даже выше, чем предполагала предыдущая теория, и (2) исследования, в которых отсутствуют доказательства генетической изменчивости SA для данного компонента приспособленности, не говорят об этом. наличие или отсутствие генетической вариации SA для приспособленности.
Хотя генетическая изменчивость SA для приспособленности может быть теоретически неизбежной и эмпирически распространенной (см. выше), степень, в которой она может объяснить изменчивость приспособленности, будет существенно расширена, если полиморфизмы SA обычно демонстрируют SSDR [41-42]. Это может вызвать чистое преимущество гетерозигот и создать стабильный уравновешивающий отбор по этим полиморфизмам [41-42]. Инверсия доминирования между аллельными вариантами в локусах, проявляющих антагонистическую плейотропию [43], была встречена с ранним скептицизмом (например,g., [44–46]), но на самом деле ожидается для среднего полиморфизма SA [47]. Кроме того, теория предсказывает, что полиморфизмы SA благоприятствуют эволюции механизмов, обеспечивающих SSDR [48]. В поддержку этого представления один из наиболее убедительных случаев специфического локуса SA, хотя и не для приспособленности как таковой, действительно демонстрирует SSDR [39].
Это может вызвать чистое преимущество гетерозигот и создать стабильный уравновешивающий отбор по этим полиморфизмам [41-42]. Инверсия доминирования между аллельными вариантами в локусах, проявляющих антагонистическую плейотропию [43], была встречена с ранним скептицизмом (например,g., [44–46]), но на самом деле ожидается для среднего полиморфизма SA [47]. Кроме того, теория предсказывает, что полиморфизмы SA благоприятствуют эволюции механизмов, обеспечивающих SSDR [48]. В поддержку этого представления один из наиболее убедительных случаев специфического локуса SA, хотя и не для приспособленности как таковой, действительно демонстрирует SSDR [39].
Наши результаты согласуются с полигенным SSDR для аллельной вариации SA, лежащей в основе приспособленности. Использование диаллельного скрещивания изогенных штаммов из хорошо охарактеризованной популяции C . maculatus , который, как известно, проявляет преимущественно SA генетическую изменчивость в приспособленности [37–38, 53, 71–72], мы показываем, что доминантно-рецессивное отношение между альтернативными аллелями в локусах, лежащих в основе приспособленности, было обратным у гетерозиготных самцов по сравнению с самками (рис. 3). ). В частности, линии, у которых значения приспособленности ауткроссированных самцов имели тенденцию к ковариации со значениями приспособленности инбредных самцов линий, с которыми они были скрещены, как правило, не проявляли этой ковариации в отношении приспособленности самок, и наоборот (см. Материалы и методы и S6 Fig).Другими словами, линии, ауткроссированная приспособленность самцов которых определялась линиями, с которыми они скрещивались (указывая на то, что аллельные вариации этих других линий были доминантными по сравнению с их собственными в отношении приспособленности самцов), как правило, были детерминантой приспособленности самок при ауткроссинге (указывая на их собственную аллельная изменчивость была доминантной по сравнению с другими штаммами в отношении приспособленности самок). Таким образом, имеет ли фиксированная аллельная вариация пригодности в данном генотипе тенденцию быть доминантной или рецессивной по отношению к вариации других генотипов (в гетерозиготном потомстве от скрещиваний между ними), зависело от пола, в котором она экспрессировалась.
3). ). В частности, линии, у которых значения приспособленности ауткроссированных самцов имели тенденцию к ковариации со значениями приспособленности инбредных самцов линий, с которыми они были скрещены, как правило, не проявляли этой ковариации в отношении приспособленности самок, и наоборот (см. Материалы и методы и S6 Fig).Другими словами, линии, ауткроссированная приспособленность самцов которых определялась линиями, с которыми они скрещивались (указывая на то, что аллельные вариации этих других линий были доминантными по сравнению с их собственными в отношении приспособленности самцов), как правило, были детерминантой приспособленности самок при ауткроссинге (указывая на их собственную аллельная изменчивость была доминантной по сравнению с другими штаммами в отношении приспособленности самок). Таким образом, имеет ли фиксированная аллельная вариация пригодности в данном генотипе тенденцию быть доминантной или рецессивной по отношению к вариации других генотипов (в гетерозиготном потомстве от скрещиваний между ними), зависело от пола, в котором она экспрессировалась. Такой SSDR будет способствовать стабильному поддержанию полиморфизмов SA для приспособленности посредством уравновешивающего отбора, увеличивая их вклад в генетическую изменчивость популяции. Действительно, это, вероятно, основной вклад в тот факт, что эта популяция демонстрирует преимущественно SA генетическую изменчивость в приспособленности [37]. Мы повторили наш тест SSDR после учета эффектов SC в данных, чтобы обеспечить более явный тест SSDR для оставшихся аллельных эффектов SA как таковых. Результат оставался очень значимым, но был немного слабее (рис. S1), что может свидетельствовать о некоторой SSDR для аллельной изменчивости с эффектами приспособленности SC (см. Введение).
Такой SSDR будет способствовать стабильному поддержанию полиморфизмов SA для приспособленности посредством уравновешивающего отбора, увеличивая их вклад в генетическую изменчивость популяции. Действительно, это, вероятно, основной вклад в тот факт, что эта популяция демонстрирует преимущественно SA генетическую изменчивость в приспособленности [37]. Мы повторили наш тест SSDR после учета эффектов SC в данных, чтобы обеспечить более явный тест SSDR для оставшихся аллельных эффектов SA как таковых. Результат оставался очень значимым, но был немного слабее (рис. S1), что может свидетельствовать о некоторой SSDR для аллельной изменчивости с эффектами приспособленности SC (см. Введение).
Предсказание о том, что отклонения доминирования (BLUP для дисперсии доминирования) должны иметь отрицательную корреляцию между признаками/полами при антагонистической плейотропии [4], не подтвердилось нашими данными (не сообщается). Другие подходы, использующие отклонения доминирования (например, корреляция аддитивной племенной ценности и отклонений доминирования), также оказались неэффективными при выявлении явного сильного признака SSDR в наших данных (не сообщается). Наконец, даже наша обширная геометрическая интерпретация компонентов дисперсии, показывающая, что дисперсия доминирования по признаку пола связана с отклонениями доминирования SA, дала неадекватную картину истинной степени SSDR, проявляемой этой популяцией.Таким образом, разделение дисперсии и корреляция отклонений доминирования, вероятно, недостаточны для документирования инверсии доминирования. Будущие исследования, направленные на изучение реверсии доминирования с помощью количественных генетических методов, должны быть направлены на проведение полных диаллельных скрещиваний и оценку относительного количества доминантных аллелей в каждом штамме с помощью метода Хеймана [50,59] (см. Материалы и методы), чтобы сопоставить эти показатели между признаки, ниши и полы, которые, как предполагается, подвергаются антагонистическому отбору.
Наконец, даже наша обширная геометрическая интерпретация компонентов дисперсии, показывающая, что дисперсия доминирования по признаку пола связана с отклонениями доминирования SA, дала неадекватную картину истинной степени SSDR, проявляемой этой популяцией.Таким образом, разделение дисперсии и корреляция отклонений доминирования, вероятно, недостаточны для документирования инверсии доминирования. Будущие исследования, направленные на изучение реверсии доминирования с помощью количественных генетических методов, должны быть направлены на проведение полных диаллельных скрещиваний и оценку относительного количества доминантных аллелей в каждом штамме с помощью метода Хеймана [50,59] (см. Материалы и методы), чтобы сопоставить эти показатели между признаки, ниши и полы, которые, как предполагается, подвергаются антагонистическому отбору.
Помимо реверсии доминирования, теория показала, что эпистатические взаимодействия между локусами SA могут способствовать поддержанию полиморфизмов SA для приспособленности [73]. Эпистаз — это ожидание полигенных признаков, основанное на геометрической модели Фишера [1,73–75]. То есть из-за уменьшающейся отдачи от дополнительных полезных аллелей во все более благоприятном фоне эффект данного аллеля, лежащего в основе непрерывного признака, зависит от того, какие аллели присутствуют в оставшихся или фоновых локусах, лежащих в основе этого признака [73–75].Несмотря на то, что это трудно обнаружить эмпирически [74] и иногда анализировать неправильно [76], существуют доказательства эпистаза с убывающей отдачей (например, [77]). Однако роль эпистаза в поддержании SA-полиморфизмов для приспособленности почти не привлекала эмпирического внимания (например, [78]). Мы обнаружили, что компонентом дисперсии, объясняющим следующую наиболее фенотипическую изменчивость в приспособленности после доминирования и доминирования SA, был эпистаз SC. Его специфический для пола аналог был идентифицирован как описывающий эпистатические эффекты SA (хотя его стандартная ошибка перекрывала ноль).
Эпистаз — это ожидание полигенных признаков, основанное на геометрической модели Фишера [1,73–75]. То есть из-за уменьшающейся отдачи от дополнительных полезных аллелей во все более благоприятном фоне эффект данного аллеля, лежащего в основе непрерывного признака, зависит от того, какие аллели присутствуют в оставшихся или фоновых локусах, лежащих в основе этого признака [73–75].Несмотря на то, что это трудно обнаружить эмпирически [74] и иногда анализировать неправильно [76], существуют доказательства эпистаза с убывающей отдачей (например, [77]). Однако роль эпистаза в поддержании SA-полиморфизмов для приспособленности почти не привлекала эмпирического внимания (например, [78]). Мы обнаружили, что компонентом дисперсии, объясняющим следующую наиболее фенотипическую изменчивость в приспособленности после доминирования и доминирования SA, был эпистаз SC. Его специфический для пола аналог был идентифицирован как описывающий эпистатические эффекты SA (хотя его стандартная ошибка перекрывала ноль). Это представляло собой одно из немногих несоответствий между нашим REML и байесовским анализом, последний выявил значительный вклад в дисперсию приспособленности от эпистатических эффектов как SC, так и SA (S3 Fig и S1 Table). Однако интерпретация эпистатических отклонений СА менее проста, чем интерпретация отклонений доминирования СА (см. выше). Во-первых, (специфические для пола) эпистатические отклонения являются свойством скрещиваний, а не штаммов, но геометрическая интерпретация этого класса наследования была основана на средних значениях деформации для (специфических для пола) эпистатических отклонений (см. Материалы и методы).Таким образом, именно скрещивания некоторых штаммов имеют тенденцию проявлять эпистатические отклонения (т. е. отклонения от ожидаемого, основанные на аддитивном вкладе каждого родительского штамма) у одного пола, но не у другого, или в разной степени у полов. Во-вторых, (SA) эпистатическая дисперсия может быть вызвана множеством взаимодействий между локусами, а не просто из-за уменьшающейся отдачи аддитивных локусов.
Это представляло собой одно из немногих несоответствий между нашим REML и байесовским анализом, последний выявил значительный вклад в дисперсию приспособленности от эпистатических эффектов как SC, так и SA (S3 Fig и S1 Table). Однако интерпретация эпистатических отклонений СА менее проста, чем интерпретация отклонений доминирования СА (см. выше). Во-первых, (специфические для пола) эпистатические отклонения являются свойством скрещиваний, а не штаммов, но геометрическая интерпретация этого класса наследования была основана на средних значениях деформации для (специфических для пола) эпистатических отклонений (см. Материалы и методы).Таким образом, именно скрещивания некоторых штаммов имеют тенденцию проявлять эпистатические отклонения (т. е. отклонения от ожидаемого, основанные на аддитивном вкладе каждого родительского штамма) у одного пола, но не у другого, или в разной степени у полов. Во-вторых, (SA) эпистатическая дисперсия может быть вызвана множеством взаимодействий между локусами, а не просто из-за уменьшающейся отдачи аддитивных локусов. Таким образом, остается неясным, являются ли обнаруженные эпистатические эффекты SA явно тем типом, который может поддерживать генетическую изменчивость приспособленности.Таким образом, в лучшем случае наши результаты могут предоставить только смешанные доказательства предполагаемой роли эпистаза убывающей отдачи SA среди локусов, лежащих в основе приспособленности в этой популяции.
Таким образом, остается неясным, являются ли обнаруженные эпистатические эффекты SA явно тем типом, который может поддерживать генетическую изменчивость приспособленности.Таким образом, в лучшем случае наши результаты могут предоставить только смешанные доказательства предполагаемой роли эпистаза убывающей отдачи SA среди локусов, лежащих в основе приспособленности в этой популяции.
Как и в случае инверсии доминирования, родительские эффекты, такие как сцепление с полом, цитоплазматические или эпигенетические эффекты, также могут частично разрешать полиморфизмы SA и способствовать поддержанию генетической изменчивости SA в приспособленности [25,79-83]. Этому есть некоторое эмпирическое подтверждение (например, [32]), но мы обнаружили небольшую или отсутствующую изменчивость в приспособленности, связанную с какой-либо формой родительских эффектов или асимметричным эпистазом (т.э., эпистаз родительских эффектов), несмотря на непревзойденное явное выявление дисперсии родительских эффектов посредством взаимных скрещиваний полного диаллеля [50]. Хотя это не означает, что родительские эффекты отсутствуют в этой популяции, это предполагает, что такие эффекты играют относительно незначительную роль в формировании дисперсии приспособленности.
Хотя это не означает, что родительские эффекты отсутствуют в этой популяции, это предполагает, что такие эффекты играют относительно незначительную роль в формировании дисперсии приспособленности.
Наше исследование предоставляет новые доказательства полигенной SSDR для генетической вариации SA, лежащей в основе приспособленности, добавляя важные сведения к нашему пониманию генетической изменчивости SA и поддержанию генетической изменчивости в приспособленности.Мы надеемся, что наши результаты будут стимулировать дальнейшие усилия в этом направлении, которые, как мы подозреваем, дополнят растущий консенсус в отношении того, что отбор SA является широко распространенным явлением среди видов, размножающихся половым путем, который обычно действует для поддержания генетической изменчивости в приспособленности.
Материалы и методы
Исследуемый организм
С . maculatus (Coleoptera: Bruchidae) — вредитель бобовых культур, колонизировавший большую часть тропических и субтропических регионов мира [84]. Таким образом, лабораторные условия (см. ниже) очень напоминают зернохранилища и посевные поля, в которых они обитали с раннего голоцена. Самки откладывают яйца на поверхность сухих бобов, а вылупившиеся личинки проникают в бобы, где завершают свой жизненный цикл, выходя из бобов в репродуктивно зрелом возрасте [84]. Этот вид является факультативным афагом (не требующим ни пищи, ни воды для успешного размножения) и демонстрирует полиандрическую систему спаривания [54], детерминацию пола X/Y [55], выраженный половой диморфизм [33,38] и экспрессию генов, предвзятую к полу [54]. 56].
Таким образом, лабораторные условия (см. ниже) очень напоминают зернохранилища и посевные поля, в которых они обитали с раннего голоцена. Самки откладывают яйца на поверхность сухих бобов, а вылупившиеся личинки проникают в бобы, где завершают свой жизненный цикл, выходя из бобов в репродуктивно зрелом возрасте [84]. Этот вид является факультативным афагом (не требующим ни пищи, ни воды для успешного размножения) и демонстрирует полиандрическую систему спаривания [54], детерминацию пола X/Y [55], выраженный половой диморфизм [33,38] и экспрессию генов, предвзятую к полу [54]. 56].
Исследуемая популяция
Происхождение нашей исследуемой популяции было описано Бергером и его коллегами [37] и Гришопом и коллегами [53]. Вкратце, популяция была выделена из семенных коробочек Vigna unguiculata , собранных на небольшом сельскохозяйственном поле недалеко от Ломе, Того (06°10′ северной широты, 01°13′ восточной долготы) в октябре и ноябре 2010 г. лаборатории и бобов, выделенных индивидуально. Девственные самцы и самки, вылупившиеся из этих бобов, были случайным образом объединены в пары, и каждая пара образовала изоженскую линию ( n = 41), каждая из которых, таким образом, произошла от одного материнского и одного отцовского генома.Эти изоженские линии были размножены и культивированы при размерах популяции 200–400 взрослых особей на 150 мл V . unguiculata при 29°C, относительной влажности 55% и световом режиме 12L:12D в течение примерно 12 поколений до проведения анализов пригодности для пола, проведенных Бергером и его коллегами [37]. Развитие изогенных (инбредных) штаммов из этих изоженских линий описано Гришопом и его коллегами [53] и в тексте S1. 16 инбредных штаммов, использованных в настоящем исследовании, были достаточно равномерно распределены по исходной межполовой генетической корреляции популяции по приспособленности (рис. S7) и, по-видимому, отражали непредвзятое представление постоянной генетической вариации SA по приспособленности, демонстрируемой дикой популяцией, от которой она была получена.
Девственные самцы и самки, вылупившиеся из этих бобов, были случайным образом объединены в пары, и каждая пара образовала изоженскую линию ( n = 41), каждая из которых, таким образом, произошла от одного материнского и одного отцовского генома.Эти изоженские линии были размножены и культивированы при размерах популяции 200–400 взрослых особей на 150 мл V . unguiculata при 29°C, относительной влажности 55% и световом режиме 12L:12D в течение примерно 12 поколений до проведения анализов пригодности для пола, проведенных Бергером и его коллегами [37]. Развитие изогенных (инбредных) штаммов из этих изоженских линий описано Гришопом и его коллегами [53] и в тексте S1. 16 инбредных штаммов, использованных в настоящем исследовании, были достаточно равномерно распределены по исходной межполовой генетической корреляции популяции по приспособленности (рис. S7) и, по-видимому, отражали непредвзятое представление постоянной генетической вариации SA по приспособленности, демонстрируемой дикой популяцией, от которой она была получена. (см. результаты и текст S1).
(см. результаты и текст S1).
Диаллельный эксперимент
Мы выполнили полное диаллельное скрещивание [50] среди 16 инбредных штаммов, получив поколение F 1 из 240 возможных ауткроссированных комбинаций и 16 родительских самости. Фенотип, который мы измерили у особей F 1 , представлял собой репродуктивный успех в течение жизни в зависимости от пола (т. е. приспособленность). F 1 мужскую ( N = 1,731) и женскую ( N = 1,547) пригодность оценивали отдельно, помещая одну фокальную особь в контейнер примерно с 25 г V . unguiculata , стерильный эталонный конкурент того же пола (из беспородной базовой популяции, созданной в то же время и из той же популяции, что и изоженские линии; см. выше), и два эталонных жука противоположного пола (пол 1:1 соотношение; см. S2 рис.). Анализы помещали в инкубаторы при 29°C, относительной влажности 55% и световом режиме 12L:12D до тех пор, пока все потомство F 2 не появилось из бобов. Количество потомков F 2 , полученных в результате анализа, представляет собой фокальную (нестерильную) приспособленность особи.Сперматозоиды стерилизованных самцов все еще функционируют и оплодотворяют яйцеклетки (но зиготы нежизнеспособны), так что тесты приспособленности самцов включали пре- и посткопулятивную селекцию (подробности см. в [72]). Тесты самок также включали конкуренцию при спаривании, а также конкуренцию за субстрат для откладки яиц (бобы) и способность самок выдерживать вредные повторные попытки спаривания со стороны конкурирующих самцов, чтобы выжить и отложить яйца [38,85–86]. Эти тесты приспособленности не только включают многие аспекты естественной экологической обстановки для этих жуков, но также представляют сложные физические среды (т.э., геометрия бобов), которые могут играть важную роль в том, чтобы лабораторные анализы приспособленности отражали сложные природные условия и позволяли брачным взаимодействиям, половому отбору и сексуальным конфликтам протекать более естественно [87–88].
Количество потомков F 2 , полученных в результате анализа, представляет собой фокальную (нестерильную) приспособленность особи.Сперматозоиды стерилизованных самцов все еще функционируют и оплодотворяют яйцеклетки (но зиготы нежизнеспособны), так что тесты приспособленности самцов включали пре- и посткопулятивную селекцию (подробности см. в [72]). Тесты самок также включали конкуренцию при спаривании, а также конкуренцию за субстрат для откладки яиц (бобы) и способность самок выдерживать вредные повторные попытки спаривания со стороны конкурирующих самцов, чтобы выжить и отложить яйца [38,85–86]. Эти тесты приспособленности не только включают многие аспекты естественной экологической обстановки для этих жуков, но также представляют сложные физические среды (т.э., геометрия бобов), которые могут играть важную роль в том, чтобы лабораторные анализы приспособленности отражали сложные природные условия и позволяли брачным взаимодействиям, половому отбору и сексуальным конфликтам протекать более естественно [87–88].
Диаллельный эксперимент проводили дважды, в двух «блоках» (с репликацией клеток внутри и между блоками). В общей сложности мы провели 3278 анализов пригодности (1731 мужчина и 1547 женщин) с умеренным дисбалансом по 256 семьям и 2 повторным блокам.Дисбаланс по категориям пола, скрещивания и/или блока был, однако, неизбежен: разные скрещивания (включая инбредные самости) давали разное количество потомков F 1 в разном соотношении полов и имели разную вероятность произвести ноль потомков F 1 , предоставляя различные возможности для анализа пригодности F 1 на протяжении всего диаллеля. Однако из-за больших усилий по выборке только 3 из 240 аутбредных скрещиваний (и 0 родительских скрещиваний) отсутствовали в общем наборе данных.
Статистическое моделирование REML
Мы подогнали пользовательскую версию модели Хеймана [59] (модифицированную для учета данных по полу), используя процедуру FDIALLEL [57] в GenStat (v.18.2.0.18409; [58]):
где μ — точка пересечения общей фенотипической дисперсии приспособленности y , которая делится на дисперсию остаточной ошибки ε , общие фиксированные эффекты повторного блока x , инбридинг b 1 , пол S и инбридинг по полу S × b 1 , а также следующие штаммо- и кросс-специфические случайные эффекты (т. д., компоненты дисперсии): аддитивная генетическая дисперсия a , доминирование b 2 , родительские эффекты c , симметричный эпистаз b 3 , асимметричный эпистаз d d случайные эффекты с S . FDIALLEL формирует факторы и матрицы, необходимые для соответствия диаллельной модели, используя директиву GenStat REML [57,89] — директива REML, допускающая другие фиксированные (например, S ) и/или случайные эффекты (например, S ).г., С × б 2 ). Модель была выполнена на данных с логарифмическим преобразованием, так как это обеспечило превосходное соответствие модели. Эта настройка подхода Хеймана [59] не изменила ни одного из лежащих в основе моделирования компонентов дисперсии, как определено в процедуре GenStat [58] FDIALLEL [57]. Мы сообщаем F-статистику и значения P для общих (фиксированных) эффектов и дисперсий ( σ 2 ) со стандартными ошибками для компонентов дисперсии (случайные эффекты).
д., компоненты дисперсии): аддитивная генетическая дисперсия a , доминирование b 2 , родительские эффекты c , симметричный эпистаз b 3 , асимметричный эпистаз d d случайные эффекты с S . FDIALLEL формирует факторы и матрицы, необходимые для соответствия диаллельной модели, используя директиву GenStat REML [57,89] — директива REML, допускающая другие фиксированные (например, S ) и/или случайные эффекты (например, S ).г., С × б 2 ). Модель была выполнена на данных с логарифмическим преобразованием, так как это обеспечило превосходное соответствие модели. Эта настройка подхода Хеймана [59] не изменила ни одного из лежащих в основе моделирования компонентов дисперсии, как определено в процедуре GenStat [58] FDIALLEL [57]. Мы сообщаем F-статистику и значения P для общих (фиксированных) эффектов и дисперсий ( σ 2 ) со стандартными ошибками для компонентов дисперсии (случайные эффекты). Компонент дисперсии с положительной дисперсией и стандартной ошибкой, исключающей ноль, интерпретируется как свидетельство того, что этот способ наследования способствует наблюдаемой фенотипической дисперсии приспособленности. При таком подходе можно получить отрицательные оценки компонента дисперсии, которые, разумеется, следует интерпретировать как не отличающиеся от нуля и не нарушающие оценки других компонентов дисперсии в модели.
Компонент дисперсии с положительной дисперсией и стандартной ошибкой, исключающей ноль, интерпретируется как свидетельство того, что этот способ наследования способствует наблюдаемой фенотипической дисперсии приспособленности. При таком подходе можно получить отрицательные оценки компонента дисперсии, которые, разумеется, следует интерпретировать как не отличающиеся от нуля и не нарушающие оценки других компонентов дисперсии в модели.
( x ) был включен в качестве дополнительного фиксированного эффекта, поскольку он имеет только два уровня — т.е.т. е. слишком мало уровней для моделирования случайного эффекта [90]. Блокировка оказала значительное влияние, вероятно, из-за дисбаланса, описанного выше — два повторных блока различались по общему размеру выборки, а также по характеру и степени дисбаланса выборки среди скрещиваний.
Для облегчения интерпретации мы также выполнили отдельные мужские и женские модели: и соответственно.
Геометрическая интерпретация классов наследования
Отдельные мужские и женские модели позволили провести геометрическую проверку значения компонентов дисперсии путем сопоставления прогнозируемых значений для каждого штамма (т. е., их BLUP) между сексированными и неполовыми моделями следующим образом. Blups для данного наследства класса Q от отдельных моделей мужской и женской ( Q M и Q F F ) Может быть установлен как дисперсионные стандартизированные y и x соответственно (рис. 1). Эту систему координат можно повернуть на 45°, чтобы получить BLUP для SC ( q SC ) и SA ( q SA ) аддитивной генетической изменчивости (рис. 1), как это сделали Бергер и его коллеги [37]. ] и Grieshop и коллеги [53], вот так:
и
Обратите внимание, что во время этой ротации дисперсия не теряется.
е., их BLUP) между сексированными и неполовыми моделями следующим образом. Blups для данного наследства класса Q от отдельных моделей мужской и женской ( Q M и Q F F ) Может быть установлен как дисперсионные стандартизированные y и x соответственно (рис. 1). Эту систему координат можно повернуть на 45°, чтобы получить BLUP для SC ( q SC ) и SA ( q SA ) аддитивной генетической изменчивости (рис. 1), как это сделали Бергер и его коллеги [37]. ] и Grieshop и коллеги [53], вот так:
и
Обратите внимание, что во время этой ротации дисперсия не теряется.
. Для многих классов наследования BLUP для q SC и q SA (полученные из отдельных мужских и женских моделей, как описано выше; см. q и S × q (оценка по полнополовой модели) соответственно. Например, в случае аддитивности a соотносились с a SC ( = 0.98, P <0,0001), а S × a коррелировало с a SA (= 0,99, P <0,0001). Таким образом, оставаясь на конкретном примере аддитивности,
и
или, говоря словами, аддитивная генетическая дисперсия a из полнополой модели представляет собой аддитивную генетическую дисперсию SC, специфичная для пола аддитивная генетическая дисперсия S × a представляет собой аддитивную генетическую дисперсию SA, и они ортогональны друг другу ( см. рис. 1).
Например, в случае аддитивности a соотносились с a SC ( = 0.98, P <0,0001), а S × a коррелировало с a SA (= 0,99, P <0,0001). Таким образом, оставаясь на конкретном примере аддитивности,
и
или, говоря словами, аддитивная генетическая дисперсия a из полнополой модели представляет собой аддитивную генетическую дисперсию SC, специфичная для пола аддитивная генетическая дисперсия S × a представляет собой аддитивную генетическую дисперсию SA, и они ортогональны друг другу ( см. рис. 1).
Убедившись, что все нераспределенные по полу компоненты дисперсии q из полнополой модели моделировали эффекты SC (см. Результаты), более прямое измерение углового соотношения между двумя осями вариации q и S × q для данный класс наследования Q (чтобы оценить, представляет ли последний дисперсию SA) является их внутренним произведением q ∙ S × q , определяемым как:
Куда Q 1 . .. Q N и S × Q 1 … S × Q N (Nonitalicized) являются блюпуми каждого штамма для дисперсионных компонентов Q и S × Q соответственно.Обратите внимание, что внутренние продукты между сексированными и неполовыми компонентами дисперсии для симметричного и асимметричного эпистаза были рассчитаны на основе средних значений деформации BLUP, поскольку эти компоненты дисперсии основаны на 120 и 240 уникальных комбинациях штамм-штамм (в диаллеле 16 × 16) соответственно. .
.. Q N и S × Q 1 … S × Q N (Nonitalicized) являются блюпуми каждого штамма для дисперсионных компонентов Q и S × Q соответственно.Обратите внимание, что внутренние продукты между сексированными и неполовыми компонентами дисперсии для симметричного и асимметричного эпистаза были рассчитаны на основе средних значений деформации BLUP, поскольку эти компоненты дисперсии основаны на 120 и 240 уникальных комбинациях штамм-штамм (в диаллеле 16 × 16) соответственно. .
Внутренний продукт между двумя осями вариации равен нулю, когда они ортогональны друг другу (или в этом случае, когда две компоненты дисперсии описывают ортогональные оси вариации). Для нормализованных осей вариации (наша стандартизирована по дисперсии до поворота системы координат; см. выше) внутренний продукт между q и S × q может быть преобразован в более интуитивно понятное угловое смещение, θ , вот так:
Таким образом, мы можем рассчитать и проверить угловое смещение всех компонентов дисперсии с разделением по полу относительно их соответствующих аналогов без пола/SC через их BLUP. Обратите внимание, что BLUP от компонентов дисперсии с отрицательными оценками (например, c и S × d ; см. Результаты) недействительны и поэтому не могут использоваться в этой геометрической интерпретации. Основное понимание, полученное в результате этого упражнения, заключается в том, что все компоненты изменчивости без пола моделируют эффекты SC и что некоторые компоненты дисперсии с разделением по полу (см. Результаты) моделируют эффекты SA в этой популяции — симметричные ортогональные отклонения от среднего значения их соответствующих аналогов без пола/SC (см. Рисунок 1).
Обратите внимание, что BLUP от компонентов дисперсии с отрицательными оценками (например, c и S × d ; см. Результаты) недействительны и поэтому не могут использоваться в этой геометрической интерпретации. Основное понимание, полученное в результате этого упражнения, заключается в том, что все компоненты изменчивости без пола моделируют эффекты SC и что некоторые компоненты дисперсии с разделением по полу (см. Результаты) моделируют эффекты SA в этой популяции — симметричные ортогональные отклонения от среднего значения их соответствующих аналогов без пола/SC (см. Рисунок 1).
Оценка SSDR
Экологическая и эпистатическая дисперсия была удалена из данных путем взятия остатков из следующей подгонки модели, снова с использованием процедуры FDIALLEL [57] в GenStat [58] (см. выше):
где μ — точка пересечения, × — фиксированный эффект повторного блока, b 3 — случайный эффект, моделирующий симметричный эпистаз, а ε — дисперсия остаточной ошибки.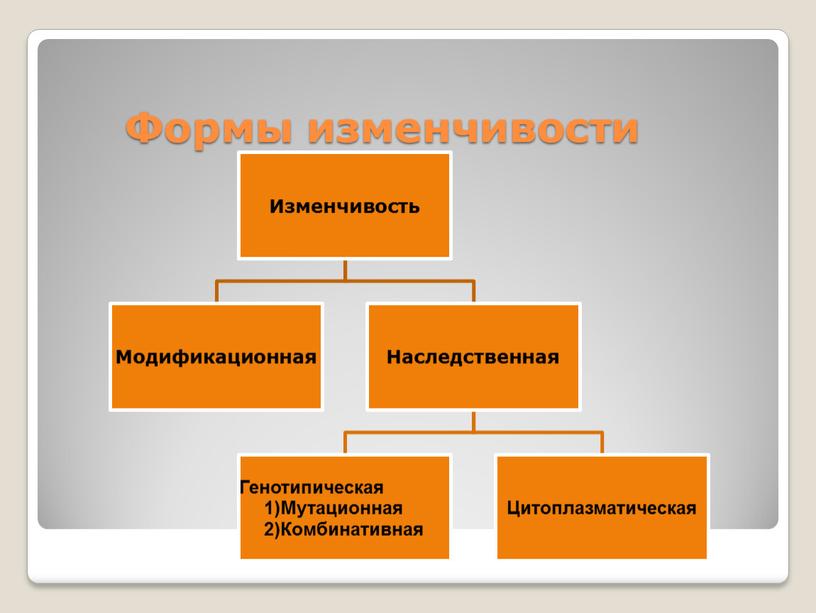 Таким образом, все другие эффекты (но именно специфические для пола аддитивные эффекты и эффекты доминирования) остаются в качестве основного вклада в дисперсию остаточных данных.Эти остатки не были стандартизованы по дисперсии.
Таким образом, все другие эффекты (но именно специфические для пола аддитивные эффекты и эффекты доминирования) остаются в качестве основного вклада в дисперсию остаточных данных.Эти остатки не были стандартизованы по дисперсии.
Средние значения семейства были сведены в таблицу из этих остатков и, σ P , r , ковариация каждой линии между средними значениями его ауткроссированного семейства ( r ) и средними значениями соответствующих инбредных родительских особей ( P ) ), которые соответствуют каждому из этих ауткроссированных семейств (т. е. W r на рис. 1 у Хеймана [59] и на рис. 20.4 у Линча и Уолша [50]), использовали в качестве оценки относительного количества рецессивные аллели в каждом штамме.Ковариации каждой линии, специфичные для производителей и маток, усредняются, и это было сделано отдельно для пригодности самцов () и самок (). Если мы обозначим семейное среднее для данного пола данной комбинации мать-отец как , то σ P , r для штамма 1 этого пола будет ковариацией элементов в этих двух векторах:
усредненный с ковариацией элементов в этих двух векторах:
Опять же, это было сделано для каждого штамма и для приспособленности самцов и самок отдельно, всего для 32 независимых ковариаций (т. д., 16 и 16; см. S6 рис.).
д., 16 и 16; см. S6 рис.).
Штаммы, чьи ауткроссированные (гетерозиготные) средства ( r ) определяются (т. е. ковариация) инбредными (гомозиготными) средствами ( P ) штаммов, с которыми они скрещиваются, имеют аллели, явно рецессивные по отношению к аллелям других штаммов, в то время как штаммы, чьи гетерозиготные средние ( r ) не зависят (т. е. не коварны) от гомозиготных средних ( P ) штаммов, с которыми они скрещены, имеют аллели, которые явно доминируют над аллелями других штаммы.Таким образом, корреляция между и дает представление о том, имеет ли аллельная изменчивость среди штаммов тенденцию к одинаковому доминантно-рецессивному соотношению у обоих полов (задается положительной корреляцией) или доминантно-рецессивное отношение аллельной изменчивости между штаммами меняется на противоположное. между полами (задается отрицательной корреляцией).
Обратите внимание, что Hayman [59] и Lynch и Walsh [50] отмечают, что σ P , r и дисперсия среди ауткроссированных семей означает r (т. е. V r на рис. 1 у Хеймана [59] и на рис. 20.4 у Линча и Уолша [50]) должны идеально масштабироваться с коэффициентом регрессии, равным 1, где точка пересечения этого наклона указывает степень проявляемого доминирования. лежащими в основе локусами (например, частичное доминирование, полное доминирование или сверхдоминирование). Это, конечно, при отсутствии эпистатической изменчивости, изменчивости окружающей среды и существенной сохраняющейся гетерозиготности у инбредных штаммов. Мы удалили эпистатическую и экологическую изменчивость (см. выше), и наши инбредные штаммы, по-видимому, сохраняют небольшую оставшуюся гетерозиготность (на что указывает большой эффект инбридинга в наших данных; см. Таблицу 1 и Таблицу S1).Тем не менее, мы по-прежнему не обнаружили связи между σ P , r и (см. выше) в отношении пригодности самцов или самок, что, возможно, указывает на различные степени доминирования среди лежащих в основе локусов и/или необъяснимую экологическую или эпистатическую дисперсия остатков.
е. V r на рис. 1 у Хеймана [59] и на рис. 20.4 у Линча и Уолша [50]) должны идеально масштабироваться с коэффициентом регрессии, равным 1, где точка пересечения этого наклона указывает степень проявляемого доминирования. лежащими в основе локусами (например, частичное доминирование, полное доминирование или сверхдоминирование). Это, конечно, при отсутствии эпистатической изменчивости, изменчивости окружающей среды и существенной сохраняющейся гетерозиготности у инбредных штаммов. Мы удалили эпистатическую и экологическую изменчивость (см. выше), и наши инбредные штаммы, по-видимому, сохраняют небольшую оставшуюся гетерозиготность (на что указывает большой эффект инбридинга в наших данных; см. Таблицу 1 и Таблицу S1).Тем не менее, мы по-прежнему не обнаружили связи между σ P , r и (см. выше) в отношении пригодности самцов или самок, что, возможно, указывает на различные степени доминирования среди лежащих в основе локусов и/или необъяснимую экологическую или эпистатическую дисперсия остатков.
Этот тест был повторен после удаления из данных аддитивных генетических эффектов SC ( a ) путем взятия остатков из следующей модели: и применение той же процедуры, описанной выше, которая обеспечивает более явный тест SSDR для генетической вариации SA как таковой.
Вспомогательная информация
S1 Рис. SSDR аллельной вариации SA, лежащей в основе приспособленности.
Диаграммы рассеяния, иллюстрирующие наблюдаемую SSDR для аллельной вариации SA, лежащей в основе приспособленности в этой популяции (т. е. в остальном идентичны рис. 3, за исключением того, что аддитивные генетические эффекты SC были заранее статистически удалены): (A) относительная величина рецессивной аллельной вариации для приспособленности у мужчин () и женщин () была значительно отрицательно коррелирована (= -0,665 [95% ДИ -0.от 87 до -0,25], P = 0,005) для разных штаммов ( N = 16; единицы отражают нестандартизированную остаточную приспособленность из модели, в которой исключена экологическая, эпистатическая и аддитивная генетическая изменчивость), и (B) та же взаимосвязь проиллюстрирована и проанализированы как ранги (т. е. штаммы ранжированы в порядке их относительного преобладания друг над другом; : -0,635, P = 0,0098). Штаммы, как правило, были обогащены SA аллельными вариациями приспособленности, которые были доминантными у их гетерозиготных сыновей, но рецессивными у их гетерозиготных дочерей, и наоборот.SA, сексуально антагонистический; SC, сексуально конкордантный; SSDR, инверсия доминирования по половому признаку.
е. штаммы ранжированы в порядке их относительного преобладания друг над другом; : -0,635, P = 0,0098). Штаммы, как правило, были обогащены SA аллельными вариациями приспособленности, которые были доминантными у их гетерозиготных сыновей, но рецессивными у их гетерозиготных дочерей, и наоборот.SA, сексуально антагонистический; SC, сексуально конкордантный; SSDR, инверсия доминирования по половому признаку.
10.1371/journal.pbio.2006810.s001
(TIF)
S2 Рис. Схема экспериментального дизайна.
Приспособленность самцов (вверху справа) и самок (внизу справа) была проанализирована в F 1 особей от всех скрещиваний (показано здесь как полученное из примера скрещивания линии 2 [как мать] и линии 3 [как отца]) и измеряли как общее количество F 2 потомков, появившихся в результате этих анализов, т.е.е., конкурентный пожизненный репродуктивный успех F 1 особей.
10.1371/journal.pbio.2006810.s002
(TIF)
S4 Рис.
 Диаллельные графики относительной пригодности.
Диаллельные графики относительной пригодности. Различие между мужскими (A) и женскими (B) апостериорными прогностическими средними значениями относительной приспособленности (т. е. приспособленности, деленной на среднюю приспособленность при ауткроссе для каждого пола) для более легкого выявления закономерностей наследования среди ауткроссированных семей (эквивалент см. на рис. S5). рисунок на логарифмически преобразованных данных, что соответствует таблице 1).Штаммы расположены в обратном порядке их средних значений HPD для аддитивной генетической изменчивости SA ( a S ), где штамм 1 является наиболее полезным для самцов/вредным для самцов, а штамм 16 является наиболее полезным/вредным для самцов. Выраженные аддитивные эффекты SC ( a ) будут представлены штаммами, имеющими относительно легко идентифицируемые вертикальные столбцы (аддитивный вклад штамма в качестве производителя) и горизонтальные ряды (аддитивный вклад штамма в качестве плотины) с постоянным оттенком, который не меняется ( много) между самцами и самками или с участием других штаммов (например,г. , штамм 9, панели А и Б). Выраженные аддитивные эффекты SA ( a S ) будут представлены легко идентифицируемыми паттернами a у одного пола данного штамма с оттенком в сторону противоположной крайности у противоположного пола этого штамма (например, штамм 1, панель А по сравнению с В). В качестве альтернативы, a S можно визуализировать, взглянув на всю популяцию: тонкий градиент от светлого к темному и от темного к светлому от верхнего левого угла к нижнему правому среди ауткроссированных семейств очевиден у самцов (A ) и самок (B), соответственно, поскольку штаммы расположены в обратном порядке их средних значений HPD для a S .Нарушения «гладкости» этого градиента, создающие более мозаичный паттерн, представляют собой основу различий в различных формах доминирования и эпистаза: b , v и w . Хотя было обнаружено, что родительские эффекты ( c ) не вносят существенного вклада в изменчивость приспособленности в этой популяции, они, в принципе, должны проявляться как различия между специфическими для отца и матери моделями a .
, штамм 9, панели А и Б). Выраженные аддитивные эффекты SA ( a S ) будут представлены легко идентифицируемыми паттернами a у одного пола данного штамма с оттенком в сторону противоположной крайности у противоположного пола этого штамма (например, штамм 1, панель А по сравнению с В). В качестве альтернативы, a S можно визуализировать, взглянув на всю популяцию: тонкий градиент от светлого к темному и от темного к светлому от верхнего левого угла к нижнему правому среди ауткроссированных семейств очевиден у самцов (A ) и самок (B), соответственно, поскольку штаммы расположены в обратном порядке их средних значений HPD для a S .Нарушения «гладкости» этого градиента, создающие более мозаичный паттерн, представляют собой основу различий в различных формах доминирования и эпистаза: b , v и w . Хотя было обнаружено, что родительские эффекты ( c ) не вносят существенного вклада в изменчивость приспособленности в этой популяции, они, в принципе, должны проявляться как различия между специфическими для отца и матери моделями a . Варианты SA любого эффекта, в принципе, выглядели бы как его аналог SC, демонстрирующий противоположный градиент оттенка между панелями A и B.HPD, самая высокая задняя плотность; SA, сексуально антагонистический; SC, сексуально конкордантный.
Варианты SA любого эффекта, в принципе, выглядели бы как его аналог SC, демонстрирующий противоположный градиент оттенка между панелями A и B.HPD, самая высокая задняя плотность; SA, сексуально антагонистический; SC, сексуально конкордантный.
10.1371/journal.pbio.2006810.s004
(TIF)
S5 Рис. Диаллельные графики логарифмической пригодности.
Разница между апостериорными прогностическими средними значениями логарифмической пригодности самцов (A) и самок (B) (отражающая анализ, представленный в таблице 1), но в остальном идентична рис. S4. полезно для визуализации фиксированных эффектов S , β и β S .Сходный средний оттенок гетерозигот между панелями А и В свидетельствует об отсутствии различий в средней приспособленности между самцами и самками ( S ). Относительная разница оттенков между инбредными родительскими «я» (по диагонали) и аутбредными гетерозиготами представляет собой эффект инбридинга ( β ), а разница в β между панелями представляет специфический для пола эффект инбридинга ( β S ), который был сильнее у самцов. Эффекты инбридинга затрудняют определение моделей наследования среди ауткроссированных семей — это легче увидеть, используя относительную пригодность (см. S4 рис.).
Эффекты инбридинга затрудняют определение моделей наследования среди ауткроссированных семей — это легче увидеть, используя относительную пригодность (см. S4 рис.).
10.1371/journal.pbio.2006810.s005
(TIF)
S6 Рис. Диаграмма, поясняющая ковариации массива, используемые для проверки SSDR.
Показанный здесь пример относится к заданному полу штамма 1 (среднее значение семьи которого индексируется как мать×отец), и в этом случае ковариация между элементами двух векторов (1×2, 1×3, … 1 × 16) и (2 × 2, 3 × 3, … 16 × 16) — соответствующие семейным средним значениям, для которых мать происходит из штамма 1, — будут усреднены с ковариацией между элементами двух векторов (2 × 1 , 3 × 1, … 16 × 1) и (2 × 2, 3 × 3, … 16 × 16) — соответствующие семейным средним значениям, для которых отец происходит из линии 1, — чтобы дать единую ковариацию для данного пола штамм 1.Это было сделано для каждой линии, для приспособленности самцов () и самок () отдельно после удаления средовой и эпистатической дисперсии из данных (рис. 3), а затем снова после удаления также аддитивных эффектов SC ( и , соответственно; S2 рис.) . SC, сексуально конкордантный; SSDR, инверсия доминирования по половому признаку.
3), а затем снова после удаления также аддитивных эффектов SC ( и , соответственно; S2 рис.) . SC, сексуально конкордантный; SSDR, инверсия доминирования по половому признаку.
10.1371/journal.pbio.2006810.s006
(TIF)
S7 Рис. Распределение изогенных штаммов относительно исходной межполовой генетической корреляции по приспособленности.
Логарифмически преобразованное и стандартизованное по дисперсии среднее значение пригодности самцов и самок для изоженских линий Бергера и его коллег [37].Закрашены и обведены красным цветом предковые изоженские линии, от которых произошли 16 изогенных штаммов настоящего исследования [53], демонстрируя, что происхождение инбредных штаммов достаточно равномерно распределено относительно исходной межполовой генетической корреляции по приспособленности. Данные, лежащие в основе этого рисунка, можно найти в цифровом репозитории Dryad, doi:10.5061/dryad.m06s2.
10.1371/journal.pbio.2006810.s007
(TIF)
S1 Таблица.
 Разбиение по байесовской дисперсии.
Разбиение по байесовской дисперсии. Байесовские диаллельные вариации (см. текст S1) для пригодности из полнополой байесовской модели для общих (фиксированных) и штаммовых и кросс-специфических (случайных) эффектов, с верхним и нижним 95% интервалами достоверности, процент объясненной дисперсии, относящейся к каждому эффект и MIP (см. Текст S1). MIP, вероятность включения модели; VarP, проекция дисперсии.
10.1371/journal.pbio.2006810.s008
(PDF)
S2 Таблица. Разделите мужские и женские байесовские модели.
BayesDiallel VarPs (см. текст S1) для отдельных мужских и женских байесовских моделей, для общих (фиксированных) и штаммовых и кросс-специфических (случайных) эффектов, с верхним и нижним 95% интервалами достоверности и процентом объясненной дисперсии, относящейся к каждой эффект.VarP, проекция дисперсии.
10.1371/journal.pbio.2006810.s009
(PDF)
S3 Таблица. Отдельные мужские и женские модели REML.
Результаты отдельных мужских и женских моделей REML, отображающих F-статистику с значениями P для общих (фиксированных) эффектов и дисперсий ( σ 2 ) с s. e. для штаммо- и кросс-специфических (случайных) эффектов (компоненты дисперсии). REML, ограниченная максимальная вероятность; с., стандартная ошибка.
e. для штаммо- и кросс-специфических (случайных) эффектов (компоненты дисперсии). REML, ограниченная максимальная вероятность; с., стандартная ошибка.
10.1371/journal.pbio.2006810.s010
(PDF)
S1 Текст. Расширенные результаты и расширенные методы.
Представление результатов байесовского анализа (и его сравнение с результатами REML) и дополнительной методологической информации об исследуемой совокупности, статистическом обосновании, байесовском статистическом моделировании, а также терминологии и значении компонентов дисперсии. REML, ограниченная максимальная вероятность.
10.1371/journal.pbio.2006810.s011
(DOCX)
Данные S1.Необработанные данные по номеру телефона и сводные данные.
Пять листов данных, описанных ниже. Необработанные данные: полные диаллельные данные, отражающие «приспособленность» особей каждого «пола» (1 = самка, 2 = самец) по отношению к идентификаторам штаммов их отцов и матерей («отцов» и «матерей» соответственно) и повторяющийся «блок», к которому принадлежит каждое наблюдение. Рис. 2: сводные данные для рис. 2, изображающие каждый компонент дисперсии («var.comp»), его «дисперсию», s.e. вокруг этой дисперсии и «верхней» и «нижней» границы этой стандартной ошибки.Рис. 3A и 3B: сводные данные для рис. 3A и 3B, изображающие идентификаторы «штаммов», ковариации массивов мужчин и женщин («m.cov» и «f.cov» соответственно) и ранговый порядок для этих ковариаций (« m.cov.rank» и «f.cov.rank» соответственно). S1A и S1B Fig: сводные данные для S1A и S1B Fig, изображающие идентификаторы «штаммов», ковариации массивов мужчин и женщин («m.cov» и «f.cov» соответственно) и ранговый порядок для этих ковариаций (« m.cov.rank» и «f.cov.rank» соответственно). S3 Fig: сводные данные для S3 Fig, изображающие каждый класс наследования («класс»), его «VarP» и «верхний» и «нижний» 95% интервалы достоверности.См. Материалы и методы. с., стандартная ошибка.
Рис. 2: сводные данные для рис. 2, изображающие каждый компонент дисперсии («var.comp»), его «дисперсию», s.e. вокруг этой дисперсии и «верхней» и «нижней» границы этой стандартной ошибки.Рис. 3A и 3B: сводные данные для рис. 3A и 3B, изображающие идентификаторы «штаммов», ковариации массивов мужчин и женщин («m.cov» и «f.cov» соответственно) и ранговый порядок для этих ковариаций (« m.cov.rank» и «f.cov.rank» соответственно). S1A и S1B Fig: сводные данные для S1A и S1B Fig, изображающие идентификаторы «штаммов», ковариации массивов мужчин и женщин («m.cov» и «f.cov» соответственно) и ранговый порядок для этих ковариаций (« m.cov.rank» и «f.cov.rank» соответственно). S3 Fig: сводные данные для S3 Fig, изображающие каждый класс наследования («класс»), его «VarP» и «верхний» и «нижний» 95% интервалы достоверности.См. Материалы и методы. с., стандартная ошибка.
10.1371/journal.pbio.2006810.s012
(XLSX)
Благодарности
Мы благодарим Дэвида Бергера, Шимона Дробняка, Сильвена Глемина, Яцека Радвана и Клауса Рюффлера за очень полезные беседы; Rafael Augusto, Mengjie Fei и Johanna Liljestrand Rönn за неоценимую техническую поддержку лаборатории; Бо Стенерлеву за доступ к установкам для облучения; Изабель Глито за коллекцию жуков; Paul Maurizio за совет с BayesDialel; и Roger Payne за поддержку FDIALLEL.
Каталожные номера
- 1. Фишер РА. Генетическая теория естественного отбора: полное издание Variorum. Издательство Оксфордского университета; 1930.
- 2. Левонтин РК. Генетическая основа эволюционных изменений. Нью-Йорк: издательство Колумбийского университета; 1974 июнь
- 3. Добжанский Т. Обзор некоторых фундаментальных понятий и проблем популяционной генетики. Cold Spring Harb Symp Quant Biol. 1955 год; 20:1–15. пмид:13433550
- 4.Чарльзворт Б., Хьюз К.А. Поддержание генетической изменчивости признаков жизненного цикла. В: Singh RS, Krimbas CB, редакторы. Эволюционная генетика. Кембридж: Издательство Кембриджского университета; 2000. с. 369–392.
- 5. Бартон Н.Х., Кейтли П.Д. Понимание количественной генетической изменчивости. Нат Рев Жене. 2002 Январь; 3 (1): 11. пмид:11823787
- 6.
Митчелл-Олдс Т., Уиллис Дж. Х., Гольдштейн Д. Б. Какие эволюционные процессы влияют на естественную генетическую изменчивость фенотипических признаков?Нат Рев Жене.
 2007 ноябрь;8(11):845. пмид:17943192
2007 ноябрь;8(11):845. пмид:17943192 - 7. Чарльзворт Б. Причины естественной изменчивости приспособленности: данные исследований популяций дрозофилы. P Natl Acad Sci. 2015 10 февраля; 112 (6): 1662–9.
- 8. Чарльзворт Б. Наследуемость приспособленности. В: Брэдбери Дж. В., Андерссон М.Б., редакторы. Половой отбор: проверка альтернатив. Чичестер: Уайли; 1987. с. 21–40.
- 9. Хоул Д. Сравнение эволюционности и изменчивости количественных признаков.Генетика. 1992 г., 1 января; 130 (1): 195–204. пмид:1732160
- 10. Холдейн Дж.Б. Математическая теория естественного и искусственного отбора, часть V: отбор и мутация. Proc Camb Philos Soc. 1927 г., июль; 23 (7): 838–844.
- 11. Мюллер ХДж. Наш груз мутаций. Am J Hum Genet. 1950 г., июнь; 2 (2): 111. пмид:14771033
- 12.
Ланде Р. Поддержание генетической изменчивости путем мутации полигенного признака со сцепленными локусами.
 Генет Рез. 1975 г., декабрь; 26 (3): 221–35.пмид:1225762
Генет Рез. 1975 г., декабрь; 26 (3): 221–35.пмид:1225762 - 13. Кроу Дж. Ф. Мутация, средняя приспособленность и генетическая нагрузка. Oxf Surv Evol Биол. 1993; 9: 3–42.
- 14. Чжан Х.С., Ван Дж., Хилл В.Г. Влияние доминантности, лептокуртоза и плейотропии вредоносных мутаций на количественную генетическую изменчивость при балансе мутаций и отбора. Генетика. 2004 г., 1 января; 166 (1): 597–610. пмид:15020447
- 15. Ланде Р. Половой диморфизм, половой отбор и адаптация полигенных признаков. Эволюция.1980 г., 1 марта; 34 (2): 292–305. пмид:28563426
- 16. Арнквист Г., Роу Л. Сексуальный конфликт. Издательство Принстонского университета; 2005.
- 17. Бондурянский Р., Ченовет С.Ф. Внутрилокусный половой конфликт. Тенденции Экол Эвол. 2009 г., 1 мая; 24 (5): 280–8. пмид:19307043
- 18.
Ван Дорн ГС. Внутрилокусный половой конфликт. Энн NY Acad Sci. 2009 г., 1 июня; 1168 (1): 52–71.

- 19. Спейер Д., Лукеш Дж., Элиаш М. Секс — вездесущий, древний и неотъемлемый атрибут эукариотической жизни.P Natl Acad Sci. 21 июля 2015 г .; 112 (29): 8827–34.
- 20. Конналлон Т., Кларк А.Г. Эволюционная неизбежность полового антагонизма. Proc Biol Sci. 2014 7 февраля; 281(1776):20132123. пмид:24335980
- 21. Конналлон Т., Кларк А.Г. Балансирующий отбор у раздельнополых видов: выводы из геометрической модели Фишера. Генетика. 1 июля 2014 г .; 197 (3): 991–1006. пмид:24812306
- 22. Кокко Х, Дженнионс, доктор медицины. Связь между половым отбором и сексуальным конфликтом.Колд Спринг Харб Перспект Биол. 2014 18 июля; 6 (9): a017517. пмид:25038050
- 23.
Конналлон Т., Кларк А.Г. Антагонистические и неантагонистические модели уравновешивающего отбора: характеристика относительных временных масштабов и эффектов автостопа при частичном селективном отборе. Эволюция. 2013 1 марта; 67 (3): 908–17.
 пмид:23461340
пмид:23461340 - 24. Конналлон Т., Кларк А.Г. Общая популяционная генетическая основа для антагонистического отбора, учитывающая демографию и повторяющиеся мутации.Генетика. 1 апреля 2012 г .; 190 (4): 1477–89. пмид:22298707
- 25. Бедхомм С., Чиппиндейл А.К. Непримиримые различия: когда половой диморфизм не может разрешить половой конфликт. В: Fairbairn DJ, Blanckenhorn W U, Székely T, редакторы. Пол, размер и гендерные роли: эволюционные исследования полового диморфизма размеров. Нью-Йорк: Издательство Оксфордского университета; 2007. с. 185–194.
- 26. Конналлон Т., Кокс Р.М., Калсбик Р. Последствия отбора по половому признаку для пригодности. Эволюция. 2010 1 июня; 64 (6): 1671–82.пмид:20050912
- 27.
Харано Т., Окада К., Накаяма С., Миятакэ Т., Хоскен Д.Дж. Внутрилокусный сексуальный конфликт, не разрешенный за счет проявления признака, ограниченного полом. Карр Биол. 2010 23 ноября; 20 (22): 2036–2039.
 пмид:21055943
пмид:21055943 - 28. Стюарт А.Д., Пишедда А., Райс В.Р. Разрешение внутрилокусного сексуального конфликта: генетические механизмы и временные рамки. Дж. Херед. 1 марта 2010 г .; 101 (дополнение 1): S94–9.
- 29. Гриффин Р.М., Дин Р., Грейс Дж.Л., Райден П., Фриберг У. Общий геном является всепроникающим ограничением эволюции экспрессии генов, зависящих от пола.Мол Биол Эвол. 2013 г., 2 августа; 30 (9): 2168–76. пмид:23813981
- 30. Федорка К.М., Муссо Т.А. Предвзятость спаривания самок приводит к противоречивой приспособленности потомства к определенному полу. Природа. 2004 г., май; 429 (6987): 65. пмид:15129280
- 31. Броммер Дж. Э., Киркпатрик М., Кварнстрем А., Густафссон Л. Межполовая генетическая корреляция для приспособленности на протяжении всей жизни в дикой природе и ее значение для полового отбора. ПЛОС ОДИН. 2007 г., 15 августа; 2(8):e744. пмид:17710144
- 32.
Ферстер К., Коулсон Т.
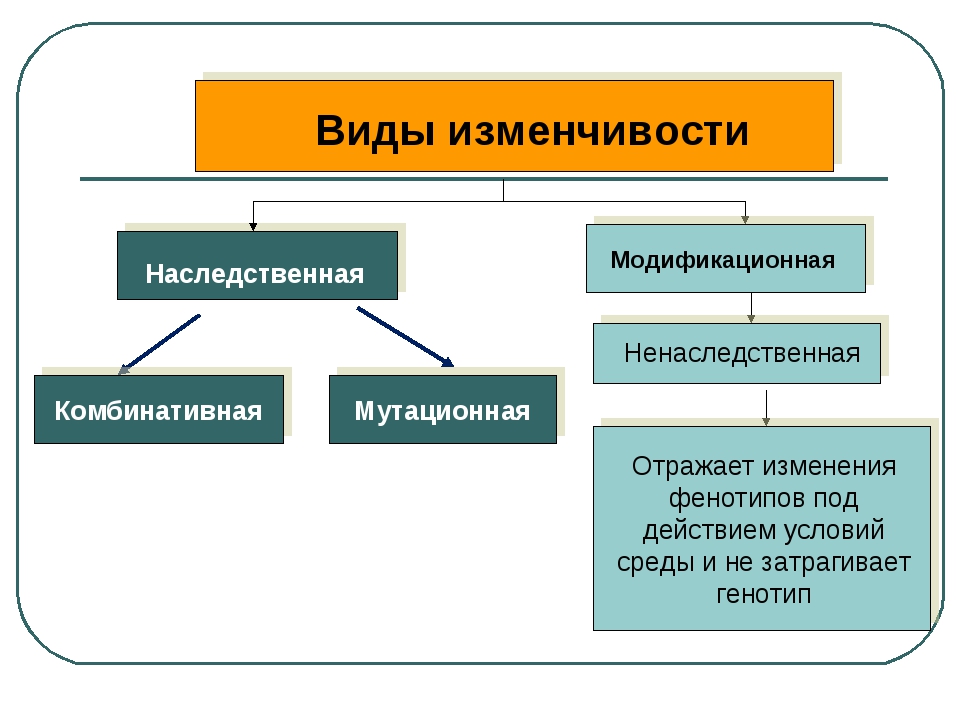 , Шелдон Б.К., Пембертон Дж.М., Клаттон-Брок Т.Х., Круук Л.Е.Антагонистические по половому признаку генетические вариации приспособленности благородных оленей. Природа. 2007 г., июнь; 447 (7148): 1107. пмид:17597758
, Шелдон Б.К., Пембертон Дж.М., Клаттон-Брок Т.Х., Круук Л.Е.Антагонистические по половому признаку генетические вариации приспособленности благородных оленей. Природа. 2007 г., июнь; 447 (7148): 1107. пмид:17597758 - 33. Арнквист Г., Туда М. Половой конфликт и гендерная нагрузка: коррелированная эволюция между приспособленностью популяции и половым диморфизмом у семенных жуков. Proc Biol Sci. 2009, 23 декабря: rspb200.
- 34. Инноченти П., Морроу Э.Х. Половые антагонистические гены Drosophila melanogaster. PLoS биол. 16 марта 2010 г .; 8 (3): e1000335. пмид:20305719
- 35.Делф Л.Ф., Андикоэчеа Дж., Стивен Дж.С., Херлихи Ч.Р., Скарпино С.В., Белл Д.Л. Зависимый от окружающей среды внутрилокусный половой конфликт у двудомного растения. Новый Фитол. 2011 1 октября; 192 (2): 542–52. пмид:21726233
- 36.
Льюис З., Уэделл Н., Хант Дж. Доказательства сильного внутрилокусного сексуального конфликта у индийской мучной моли, Plodia interpunctella.
 Эволюция. 2011 г., 1 июля; 65 (7): 2085–97. пмид:21729062
Эволюция. 2011 г., 1 июля; 65 (7): 2085–97. пмид:21729062 - 37. Бергер Д., Гришоп К., Линд М.И., Гоэнага Дж., Маклаков А.А., Арнквист Г.Внутрилокусный сексуальный конфликт и стресс окружающей среды. Эволюция. 2014 г., 1 августа; 68 (8): 2184–96. пмид:24766035
- 38. Бергер Д., Мартиносси-Аллиберт И., Гришоп К., Линд М.И., Маклаков А.А., Арнквист Г. Внутрилокусный половой конфликт и трагедия общин у семенных жуков. Я Нат. 1 октября 2016 г .; 188 (4): E98–112. пмид:27622882
- 39. Барсон Н.Дж., Айканат Т., Хиндар К., Барански М., Болстад Г.Х., Фиске П. и соавт. Зависимое от пола доминирование в одном локусе поддерживает различия в возрасте половозрелости у лосося.Природа. 2015 декабрь; 528 (7582): 405. пмид:26536110
- 40. Ченг С., Киркпатрик М. Отбор по половому признаку и экспрессия генов, обусловленная полом, у людей и мух. Генетика PLoS. 22 сентября 2016 г .; 12 (9): e1006170. пмид:27658217
- 41.
 Кидвелл Дж. Ф., Клегг М. Т., Стюарт Ф. М., Праут Т. Области стабильного равновесия для моделей дифференциального отбора у двух полов при случайном спаривании. Генетика. 1977 г., 28 января; 85 (1): 171–83. пмид:838269
Кидвелл Дж. Ф., Клегг М. Т., Стюарт Ф. М., Праут Т. Области стабильного равновесия для моделей дифференциального отбора у двух полов при случайном спаривании. Генетика. 1977 г., 28 января; 85 (1): 171–83. пмид:838269 - 42. Праут Т. Насколько хорошо противоположный отбор поддерживает изменчивость?В: Singh RS, Krimbas CB, редакторы. Эволюционная генетика. Кембридж: Издательство Кембриджского университета; 2000. с. 157–181.
- 43. Роуз МР. Антагонистическая плейотропия, доминирование и генетическая изменчивость. Наследственность. 1982 г., февраль; 48 (1): 63.
- 44. Хоэкстра Р.Ф., Бийлсма Р., Долман А.Дж. Полиморфизм из-за неоднородности окружающей среды: модели устойчивы только в том случае, если гетерозигота близка по приспособленности к предпочтительной гомозиготе в каждой среде. Генетические исследования. 1985 г., июнь; 45 (3): 299–314.
- 45.
Curtsinger JW, Service PM, Prout T. Антагонистическая плейотропия, изменение доминирования и генетический полиморфизм.
 Я Нат. 1994 г., 1 августа; 144 (2): 210–28.
Я Нат. 1994 г., 1 августа; 144 (2): 210–28. - 46. Хедрик П.В. Антагонистическая плейотропия и генетический полиморфизм: перспектива. Наследственность. 1999 г., февраль; 82 (2): 126–33.
- 47.
Фрай Джей Ди. Геномное расположение половых антагонистических вариаций: некоторые предостерегающие комментарии. Эволюция. 2010 1 мая; 64 (5): 1510–6. пмид:19
3
- 48.Спенсер Х.Г., священник Н.К. Эволюция специфического для пола доминирования в ответ на антагонистический по половому признаку отбор. Я Нат. 2016 1 мая; 187 (5): 658–66. пмид:27104997
- 49. Фальконер Д.С., Маккей Т.Ф. Введение в количественную генетику. 4-е изд. Нью-Йорк: Пирсон/Прентис Холл; 1996.
- 50. Линч М., Уолш Б. Генетика и анализ количественных признаков. Сандерленд, Массачусетс: Синауэр; 1998.
- 51.
Гарсия-Дорадо А., Кабальеро А. О среднем коэффициенте доминирования вредных спонтанных мутаций.Генетика, 2000 г., 1 августа; 155 (4): 1991–2001 гг.
 пмид:101
пмид:101 - 52. Манна Ф., Мартин Г., Ленорман Т. Фитнес-ландшафты: альтернативная теория доминирования мутаций. Генетика, 1 января 2011 г .; 189 (3): 923–937. пмид:218
- 53. Гришоп К., Бергер Д., Арнквист Г. Сексуально антагонистические генотипы с мужской выгодой демонстрируют повышенную уязвимость к инбридингу. БМС Эвол Биол. 2017 дек;17(1):134. пмид:28606137
- 54. Миятакэ Т., Мацумура Ф. Внутривидовая изменчивость повторного спаривания самок у Callosobruchus chinensis и C.пятно. Дж. Физиология насекомых. 2004 г., 1 мая; 50 (5): 403–8. пмид:15121453
- 55. Ангус Р.Б., Деллоу Дж., Уиндер С., Кредленд П.Ф. Различия кариотипов у четырех видов Callosobruchus Pic (Coleoptera: Bruchidae). Журнал исследований хранимых продуктов. 2011 1 апреля; 47 (2): 76–81.
- 56.
Иммонен Э., Саяди А., Байрам Х., Арнквист Г. Спаривание изменяет экспрессию генов полового диморфизма у семенного жука Callosobruchus maculatus.
 Геном Биол Эвол. 2017 1 марта; 9 (3): 677–99. пмид:283
Геном Биол Эвол. 2017 1 марта; 9 (3): 677–99. пмид:283 - 57.Пейн РВ. Процедура FDIALLEL В справочном руководстве Genstat, выпуск 18, часть 3, библиотека процедур PL26. [программное обеспечение]. Хемел Хемпстед: VSN International. 2015.
- 58. ВСН Интернэшнл. Genstat для Windows 18th Edition VSN International [программное обеспечение]. Хемел Хемпстед, Великобритания. 2015. Доступно на: Genstat.co.uk.
- 59. Хейман Б.И. Теория и анализ диаллельных скрещиваний. Генетика. 1954 г., ноябрь; 39 (6): 789–809. пмид:17247520
- 60. Ленарчич А.Б., Свенсон К.Л., Черчилль Г.А., Валдар В.Общий байесовский подход к анализу диаллельных скрещиваний инбредных штаммов. Генетика. 1 февраля 2012 г .; 190 (2): 413–35. пмид:22345610
- 61. Ленарчич АБ. BayesDiallel: пакет программного обеспечения BayesDiallel R. Версия 098 [программное обеспечение]. 2014.
- 62.
R Core Team R: язык и среда для статистических вычислений [программное обеспечение].
 Фонд R для статистических вычислений, Вена, Австрия. 2015. Доступно по адресу: http://www.R-project.org/.
Фонд R для статистических вычислений, Вена, Австрия. 2015. Доступно по адресу: http://www.R-project.org/. - 63. Маллет М.А., Чиппиндейл А.К.Инбридинг показывает более сильный сетевой отбор самцов Drosophila melanogaster: последствия для мутационной нагрузки и приспособленности половых самок. Наследственность. 2011 г., июнь; 106 (6): 994–1002. пмид:21119701
- 64. Манк Дж. Популяционная генетика полового конфликта в геномную эпоху. Нат Рев Жене. 2017 дек;18(12):721. пмид:257
- 65. Роу Л., Ченовет С.Ф., Агравал А.Ф. Геномика сексуального конфликта. Я Нат. 2018 1 августа; 192 (2): 274–286. пмид:30016158
- 66.Елена С, Ленский РЭ. Эволюционные эксперименты с микроорганизмами: динамика и генетические основы адаптации. Нат Рев Жене. 2003;4(6):457–469. пмид:12776215
- 67.
Qian W, Ma D, Xiao C, Wang Z, Zhang J. Геномный ландшафт и эволюционное разрешение антагонистической плейотропии у дрожжей. Сотовые отчеты.
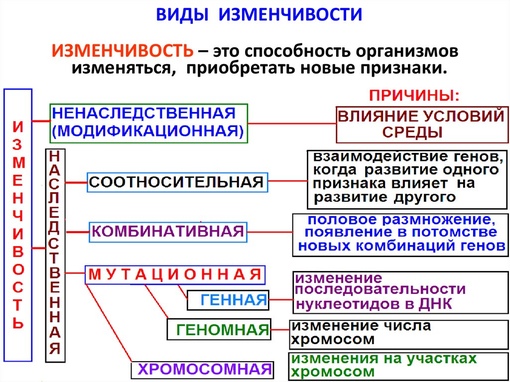 2012 г., 29 ноября; 2 (5): 1399–410. пмид:23103169
2012 г., 29 ноября; 2 (5): 1399–410. пмид:23103169 - 68. Орр ХА. Адаптация и цена сложности. Эволюция. 2000 г., февраль; 54 (1): 13–20. пмид:10937178
- 69.Павличев М, Вагнер ГП. Модель эволюции развития: отбор, плейотропия и компенсация. Тенденции Экол Эвол. 2012 1 июня; 27 (6): 316–22. пмид:22385978
- 70. Зайчек Ф., Конналлон Т. Антагонистическая плейотропия у раздельнополых видов и сохранение генетической изменчивости в жизненных признаках и приспособленности. Эволюция. 2018 17 апр.
- 71. Бергер Д., Ю Т., Минано М. Р., Гришоп К., Линд М. И., Арнквист Г., Маклаков А. А. Половой антагонистический отбор на генетической изменчивости, лежащей в основе однополого сексуального поведения как мужчин, так и женщин.БМС Эвол Биол. 2016 дек;16(1):88.
- 72.
Гришоп К., Стенгберг Дж., Мартиносси-Аллиберт И., Арнквист Г., Бергер Д. Сильный половой отбор у самцов против мутационной нагрузки, снижающей производство потомства у семенных жуков.
 Дж. Эвол Биол. 2016 1 июня; 29 (6): 1201–10. пмид:269
Дж. Эвол Биол. 2016 1 июня; 29 (6): 1201–10. пмид:269 - 73. Арнквист Г., Веллноу Н., Роу Л. Влияние эпистаза на половые антагонистические генетические вариации. Proc Biol Sci. 2014 22 июля; 281 (1787): 20140489. пмид:24870040
- 74.Whitlock MC, Phillips PC, Moore FB, Tonsor SJ. Множественные фитнес-пики и эпистазы. Annu Rev Ecol Evol Syst. 1995 ноябрь; 26 (1): 601–29.
- 75. Мартин Г., Елена С.Ф., Ленорман Т. Распределение эпистаза у микробов соответствует прогнозам модели фитнес-ландшафта. Нат Жене. 2007 г., апрель; 39 (4): 555–560. пмид:17369829
- 76. Бергер Д., Постма Э. Предвзятые оценки эпистаза с убывающей отдачей? Повторное рассмотрение эмпирических данных. Генетика. 1 декабря 2014 г .; 198 (4): 1417–20.пмид:25313131
- 77.
Хан А.И., Динь Д.М., Шнайдер Д., Ленски Р.Е., Купер Т.Ф. Отрицательный эпистаз между полезными мутациями в эволюционирующей бактериальной популяции. Наука. 2011 3 июня; 332 (6034): 1193–1196.
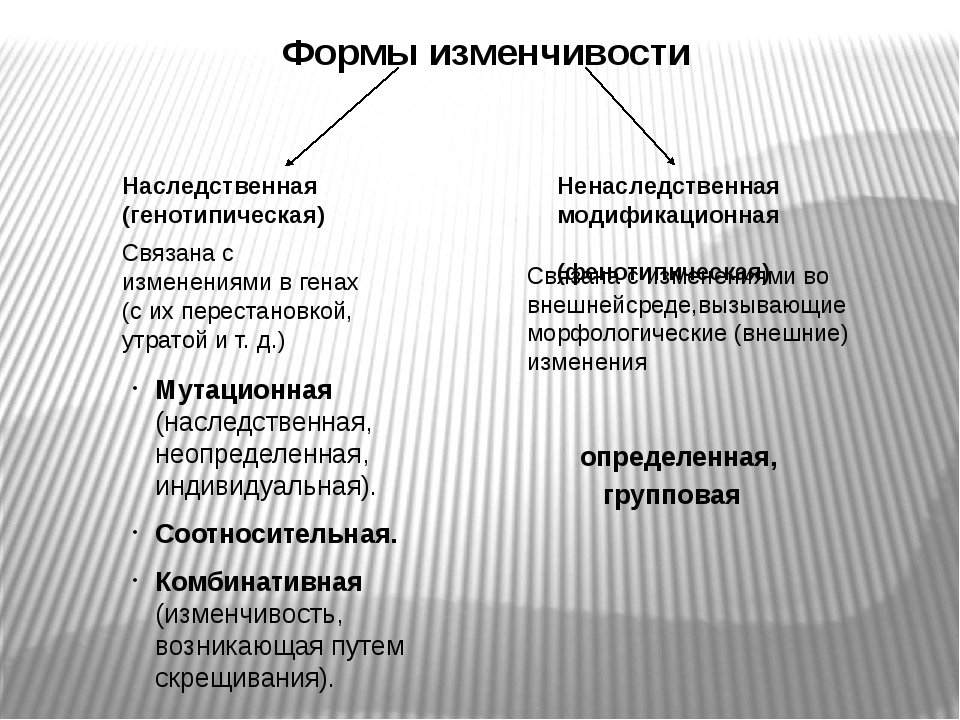 пмид:21636772
пмид:21636772 - 78. Smith DT, Hosken DJ, Rostant WG, Yeo M, Griffin RM, Bretman A, Price TA, Ffrench-Constant RH, Wedell N. Устойчивость к ДДТ, эпистаз и приспособленность самцов мух. Дж. Эвол Биол. 2011 июнь; 24 (6): 1351–62. пмид:21507117
- 79. Рис ВР.Половые хромосомы и эволюция полового диморфизма. Эволюция. 1984 г., 1 июля; 38 (4): 735–42. пмид:28555827
- 80. Ланде Р., Киркпатрик М. Реакция отбора признаков с материнским наследованием. Генетические исследования. 1990 июнь; 55 (3): 189–97.
- 81. Муссо Т.А., Фокс CW. Адаптивное значение материнских эффектов. Тенденции Экол Эвол. 1998 г., 1 октября; 13 (10): 403–7. пмид:21238360
- 82. День Т., Бондурянски Р. Сексуальный конфликт внутри локуса может стимулировать эволюцию геномного импринтинга.Генетика. 2004 г., 1 августа; 167 (4): 1537–46. пмид:15342496
- 83.
Бондурянски Р., Дэй Т. Негенетическое наследование и его эволюционные последствия.
 Annu Rev Ecol Evol Syst. 2009 1 декабря; 40: 103–25.
Annu Rev Ecol Evol Syst. 2009 1 декабря; 40: 103–25. - 84. Саутгейт БиДжей. Биология Брухид. Ежегодный обзор энтомологии. 1979 г., январь; 24 (1): 449–73.
- 85. Рённ Дж., Катвала М., Арнквист Г. Затраты на спаривание и производство яиц у жуков-семенников Callosobruchus. Аним Бехав. 2006 г., 1 августа; 72 (2): 335–42.
- 86.
Hotzy C, Arnqvist G. Конкуренция сперматозоидов благоприятствует вредным самцам у семенных жуков. Карр Биол. 10 марта 2009 г .; 19 (5): 404–7. пмид:1
65
- 87. Сингх А., Агравал А.Ф., Рандл Х.Д. Экологическая сложность и очистка вредных аллелей. Эволюция. 2017 ноябрь; 71 (11): 2714–20. пмид:28840604
- 88. Юн Л., Чен П.Дж., Сингх А., Агравал А.Ф., Рандл Х.Д. Физическая среда опосредует вред мужчин и его влияние на отбор женщин. Proc Biol Sci.2017 12 июля; 284 (1858): 20170424. пмид:28679725
- 89.
Справочное руководство VSN International Genstat (выпуск 18), часть 2 Директивы [программное обеспечение].
 VSN International, Хемел-Хемпстед, Великобритания. 2015.
VSN International, Хемел-Хемпстед, Великобритания. 2015. - 90. Болкер Б.М., Брукс М.Е., Кларк С.Дж., Джиндж С.В., Поулсен Дж.Р., Стивенс М.Х., Уайт Дж.С. Обобщенные линейные смешанные модели: практическое руководство по экологии и эволюции. Тенденции Экол Эвол. 2009 1 марта; 24 (3): 127–35. пмид:186
Влияние генетической изменчивости на экспрессию генов
- Рохан Б.Х. Уильямс1,2,3,4,
- Ева К.Ф. Чан1,3,5,
- Марк Дж. Коули1,4 и
- Питер Ф.Р. Маленький1,6
- 1 Школа биотехнологии и биомолекулярных наук Университета Нового Южного Уэльса, Рандвик, Новый Южный Уэльс, 2052, Австралия;
- 2 Центр анализа функций генов им.
 Рамачиотти, Университет Нового Южного Уэльса, Рандвик, Новый Южный Уэльс, 2052, Австралия
Рамачиотти, Университет Нового Южного Уэльса, Рандвик, Новый Южный Уэльс, 2052, Австралия
-
↵3 Эти авторы внесли одинаковый вклад в эту работу.
Аннотация
Мнение о том, что изменения в контроле экспрессии генов, а не изменения в последовательности белков, являются центральными для эволюции.
организмов стало чем-то вроде трюизма в молекулярной биологии. На самом деле прямых доказательств этого мало, и
только недавно у нас появилась возможность взглянуть более глобально на то, как генетическая изменчивость влияет на экспрессию генов, сосредоточив внимание
при межиндивидуальных вариациях экспрессии генов и использовании микрочипов для проверки различий в уровнях мРНК. Мы тут
рассмотреть масштабы этих экспериментальных анализов, что они призваны рассказать нам о генетической изменчивости и каковы их
ограничения как с технической, так и с концептуальной точки зрения. Мы заключаем, что, хотя мы начинаем понимать влияние
этого класса генетической изменчивости при стационарных уровнях мРНК, мы все еще далеки от идентификации потенциальных фенотипических
и эволюционные результаты.
Мы тут
рассмотреть масштабы этих экспериментальных анализов, что они призваны рассказать нам о генетической изменчивости и каковы их
ограничения как с технической, так и с концептуальной точки зрения. Мы заключаем, что, хотя мы начинаем понимать влияние
этого класса генетической изменчивости при стационарных уровнях мРНК, мы все еще далеки от идентификации потенциальных фенотипических
и эволюционные результаты.
Концептуальная основа
Прямой анализ стационарных уровней мРНК у особей одного и того же вида показывает, что количество мРНК может различаться
между людьми, предполагая, что генетическая изменчивость может влиять на количество мРНК в клетке.Расширение этих наблюдений
попытка установить формальную генетическую основу для некоторых из этих вариаций является областью исследований, которая первоначально
Янсен и Нап (2001) назвали ее «генетической геномикой», а в настоящее время в просторечии ее называют «экспрессионной генетикой».
Учитывая давний теоретический интерес к вариациям в контроле экспрессии генов как к движущей силе эволюции (King and Wilson 1975), прямых доказательств важности этого типа генетической изменчивости на удивление мало.Например, Беннетт и др. (1995) предположили, что вариации в ДНК рядом с генами INS и IGF2 у людей приводят к изменениям уровней мРНК, которые связаны с предрасположенностью к диабету 1 типа, а Theuns et al. (2000) предположили, что вариации в промоторной области гена PSEN1 способствуют повышенному риску раннего начала болезни Альцгеймера.
Важно понимать, что изменение количества мРНК не обязательно должно приравниваться к изменению транскрипции на
се.На рис. 1 схематически показаны множественные процессы, которые в конечном счете влияют на уровень мРНК в клетке.
вариации в любой части этого процесса в принципе могут приводить к изменениям стационарного уровня мРНК. Многие из этих процессов
управляются молекулярными машинами, содержащими от 10 до сотен компонентов (Маниатис и Рид, 2002; Масиаг и др., 2006; Цанков и др., 2006), но нам неизвестны какие-либо систематические исследования степени полиморфизма в компонентах этих машины.Мы признаем
что изменение в контроле альтернативного сплайсинга (например, см. Hull et al. 2007) может повлиять на попытки измерить стационарные уровни мРНК с помощью геноспецифических ДНК-зондов, и мы не пытались расширить
наш обзор в этой области.
Многие из этих процессов
управляются молекулярными машинами, содержащими от 10 до сотен компонентов (Маниатис и Рид, 2002; Масиаг и др., 2006; Цанков и др., 2006), но нам неизвестны какие-либо систематические исследования степени полиморфизма в компонентах этих машины.Мы признаем
что изменение в контроле альтернативного сплайсинга (например, см. Hull et al. 2007) может повлиять на попытки измерить стационарные уровни мРНК с помощью геноспецифических ДНК-зондов, и мы не пытались расширить
наш обзор в этой области.
Вероятные места действия генетических детерминант уровней мРНК.Генетические вариации, влияющие на экспрессию генов, могут
в регуляторных последовательностях, промоторах, энхансерах, сайтах сплайсинга и мотивах вторичной структуры целевого гена и
таким образом, быть генетически в цис (красные звезды), или могут быть вариации молекулярного механизма, которые взаимодействуют с цис -регуляторными последовательностями и, таким образом, действуют генетически в транс (синие звезды).
Признавая эту сложность, в этом обзоре мы будем называть все эти возможные влияния опосредованными «регуляторы» без признания их биологической роли; подчеркиваем, регулятор не обязательно должен отождествляться с фактором транскрипции.Мы также описываем все гены, находящиеся под влиянием регулятора, как «регулируемые» или «находящиеся под влиянием», опять же, независимо от фактического задействованные механизмы.
На первый взгляд, фундаментальный вопрос, на который пытаются ответить все исследования, заключается в том, в какой степени гены подвержены или не подвержены влиянию
генетической изменчивостью и характером этих влияний. К сожалению, наши знания об экспрессии генов предполагают, что это
вопрос сам по себе несколько упрощен. Экспрессия генов во многих, а может быть, и в большинстве случаев чувствительна к внешнему контролю.
таким образом, ген может подвергаться базальному влиянию, то есть при всех условиях экспрессии, или может подвергаться условному влиянию из-за
генетическая изменчивость молекулярного механизма, который контролирует уровни мРНК во время развития или в ответ на воздействие окружающей среды.
указанные изменения. В многоклеточных организмах эта проблема усугубляется потенциальным дифференцированным контролем
генов в разных клетках или тканях.Совершенно ясной целью некоторых из этих исследований является установление степени, в которой это
тип генетической изменчивости является тканеспецифичным, но важно понимать, что это только один из важных
контроля, которые применяются к генам.
Экспрессия генов во многих, а может быть, и в большинстве случаев чувствительна к внешнему контролю.
таким образом, ген может подвергаться базальному влиянию, то есть при всех условиях экспрессии, или может подвергаться условному влиянию из-за
генетическая изменчивость молекулярного механизма, который контролирует уровни мРНК во время развития или в ответ на воздействие окружающей среды.
указанные изменения. В многоклеточных организмах эта проблема усугубляется потенциальным дифференцированным контролем
генов в разных клетках или тканях.Совершенно ясной целью некоторых из этих исследований является установление степени, в которой это
тип генетической изменчивости является тканеспецифичным, но важно понимать, что это только один из важных
контроля, которые применяются к генам.
Генетика экспрессии как область деятельности сосредоточена в первую очередь на картировании генетических детерминант вариаций уровня мРНК, в основном
рассматривая уровни мРНК как непрерывно изменяющийся фенотип; они поддаются анализу как количественный признак или «QT». Поскольку мРНК
является результатом экспрессии гена, фраза «выражение количественного локуса признака» или «eQTL» была придумана для описания
этот анализ. Для тех, кто не знаком с терминологией, важно понимать, что термин eQTL явно относится
к картированному локусу, который влияет на вариабельный уровень мРНК, а не на сам признак экспрессии мРНК (QT).
Поскольку мРНК
является результатом экспрессии гена, фраза «выражение количественного локуса признака» или «eQTL» была придумана для описания
этот анализ. Для тех, кто не знаком с терминологией, важно понимать, что термин eQTL явно относится
к картированному локусу, который влияет на вариабельный уровень мРНК, а не на сам признак экспрессии мРНК (QT).
Стационарные уровни мРНК нескольких генов могут быть измерены либо с помощью микрочипов, либо, реже, путем прямого секвенирования кДНК.
(например, см. Cowles et al.2002). Обнаружение на основе ПЦР (Singer-Sam et al. 1992) экспрессии двух аллелей гена, кодирующего молчащие замены, также использовалось (например, Yan et al. 2002) для обнаружения вариаций относительных уровней мРНК, полученных из отдельных аллелей. набора генов. Микроматричный анализ был
гораздо более широко используется и имеет преимущество перед другими методами в том, что они содержат генные комплементы, близкие к глобальным; микрочип
исследования будут в центре внимания нашего обзора.
Экспериментальный план всех исследований, которые мы рассмотрим здесь, концептуально идентичен. Количество стационарной мРНК в одной или нескольких тканях или во всем организме измеряют на панели генетически типированных особей; далее разнообразие статистических подходов используются для определения того, как изменения относительного выхода мРНК в группе могут или не могут коррелировать с генетическими маркерами.Статистически значимая корреляция изменения уровня мРНК с определенными маркерами предполагает, что что маркеры определяют приблизительное местоположение вариантного регулятора, влияющего на выход мРНК.
В этом обзоре мы сначала обсудим технические трудности, связанные с анализом микрочипов в генетическом контексте; мы затем обзор основных исследований, проведенных на системах млекопитающих, со ссылкой на ключевые исследования, проведенные на дрожжах, и, наконец, мы обсуждаем основные биологические интерпретации данных экспериментов экспрессионной генетики.Обращаем внимание на недавнее обзоры Rockman and Kruglyak (2006) и Gibson and Weir (2005), а также специальный выпуск Mammalian Genome (июнь 2006 г.), который содержит несколько обзорных статей по более специализированным аспектам этой области.
Технический анализ экспериментов с микрочипами
Использование микрочипов для измерения уровней мРНК — это мощный метод, связанный с многочисленными экспериментальными артефактами. а также наличие четких и воспроизводимых результатов (обзор см. в Консорциуме контроля качества микрочипов 2006 г.).С точки зрения экспрессионной генетики микрочипы имеют определенные недостатки как в экспериментальных, так и в аналитических данных. процедуры, которые могут существенно повлиять на результат анализа экспрессионного картирования. Эти проблемы отчасти являются общими при любом анализе микрочипов, но помещенном в контекст картографических панелей, артефакт может быть формально неверно истолкован как eQTL, который изменяет уровень экспрессии гена, экспрессия которого на самом деле не является генетически изменчивой.Результат во многих случаях является ошибочной идентификацией того, какие гены связаны и какие области генома содержат eQTL.
Соответствующие исследования выявили следующее: Doss et al. (2005) и Alberts et al. (2005) продемонстрировали искусственную идентификацию цис -eQTL, возникающую из-за SNP в мРНК-мишени, которая должна быть опрошена с помощью короткого гибридизационного зонда, который, как следствие несоответствия между зондом и мишенью, приводит к уменьшению гибридизации в некоторых генетических фонах, но не другие, совершенно независимо от фактических уровней мРНК.Альбертс и др. (2005) и Уильямс и др. (2006) определили, что артефакты из-за различий в партиях между группами микрочипов могут привести к ложному мастер-регулятору. eQTL, которые представляют собой eQTL, которые, по-видимому, связаны/ассоциируются с большим количеством генов (более подробное обсуждение см. ниже). Уильямс и др. (2006) также показали, что может быть плохая согласованность между eQTL, связанными с геном, после нормализации несколькими различными, но широко используемые процедуры обработки необработанных данных микрочипов.Наконец, Мэнли и др. (2005) показали, что некоторые массивы содержат несколько зондов, которые, в принципе, представляют один и тот же ген, но эти разные зонды идентифицировали различные eQTL.
Несмотря на эти сложности, существует значительный объем исследований, сообщающих о результатах экспрессионного генетического анализа. основные исследования обсуждаются непосредственно ниже.
Обзор ключевых исследований
В таблице 1 перечислены ключевые эксперименты в масштабе всего генома, которые, по нашему мнению, представляют собой основы экспрессионной генетики.Неизбежно, между этими исследованиями есть большие экспериментальные различия, включая организмы, количество особей, их генетическая сложность (F 2 , гаплоидный, инбредный, аутбредный), источники мРНК (типы тканей/клеток), платформы микрочипов, типы и количество генетических маркеров (SNP или более длинные повторы), критерии включения уровней мРНК, которые могли применяться до анализа, методы оценки eQTL-связь/ассоциация и методы коррекции множественного тестирования и оценки значимости.В результате этих диспропорций возможно, неудивительно, что между их результатами существуют значительные несоответствия.
Таблица 1.Краткое изложение экспериментальной и аналитической методологии и результатов исследований генетики ключевой экспрессии
Принимая во внимание присущие этим исследованиям различия, мы теперь обсудим, какой вклад внесли эти исследования картирования eQTL. нашему пониманию в четырех областях: количество обнаруживаемых генетических влияний, характер генетического влияния, мастер регуляторы экспрессии генов и, наконец, множественный генетический контроль вариаций экспрессии генов.
Количество обнаруживаемых генетических влияний
Одним из наименее последовательных результатов является доля признаков экспрессии, демонстрирующих сцепление или ассоциацию по крайней мере с один eQTL, который колеблется от 0,8% до 59% уровней мРНК генов (таблица 2). Нижний предел в 0,8% взят из ассоциативного исследования (Stranger et al.2005) из 374 генов, удовлетворяющих критериям включения, что общий уровень гибридизации и дисперсия высоки в 60 образцах неродственные люди. Из этих 374 транскриптов только 0,6% были связаны с eQTL, определяемым порогом Бонферрони, равным 0,05. Напротив, верхний предел в 59% получен из исследования сцепления (Brem and Kruglyak 2005), в ходе которого был обнаружен по крайней мере один eQTL для 2984 генов, определяемый порогом частоты ложных открытий (FDR) равным 0,05. Многие из этих различия можно объяснить несхожестью экспериментальной методологии, но в среднем можно обнаружить хотя бы один eQTL для от 10% до 30% исследованных генов (см. Таблицу 2).
Таблица 2.Краткое описание характера генетического влияния на экспрессию генов
Статистическая мощность также оказывает большое влияние на количество генов, для которых может быть идентифицирован eQTL.Например, Брем и др. (2002) с использованием 40 образцов гаплоидных дрожжей определили eQTL для 9% генов при P < 5 × 10 -5 , тогда как Yvert et al. (2003) с использованием почти идентичной методологии и 86 образцов гаплоидных дрожжей определили eQTL для 40% генов при P < 3,4 × 10 -5 . Поскольку основное различие между этими двумя исследованиями заключается преимущественно в размере выборки, эти результаты предполагают, что удвоение в размере выборки потенциально может увеличить мощность обнаружения eQTL в четыре раза.Важное сообщение на вынос в выражении генетики заключается в том, что размер картографической панели является важным фактором, определяющим успех, урок, хорошо задокументированный в картографировании. сложных заболеваний человека.
Характер изменения:
цис — против транс -действующее изменениеГенетические вариации, влияющие на экспрессию генов, могут находиться в пределах регуляторных последовательностей, таких как промоторы, энхансеры, сплайсинг сайты или мотивы вторичной структуры гена-мишени, и поэтому генетически находятся в цис , или они могут быть вариациями белков и РНК, которые взаимодействуют с цис -регуляторными последовательностями и, таким образом, генетически находятся в транс (рис.1). Поскольку ген, связанный с каждым признаком экспрессии мРНК, может быть физически картирован в геноме, механизм ( цис -действующий или транс -действующий), с помощью которого eQTL влияет на признак экспрессии, может быть выведен из близости между физическим расположением родственного гена и eQTL. Этот процесс может быть спутан транс -действующими влияниями, которые случайно отображаются в соответствующем окне, которое используется для определения цис (Ronald et al.2005). Чтобы преодолеть логическую трудность, связанную с тесно сцепленным транс -регулятором, Rockman and Kruglyak (2006) различают локальное и отдаленное сцепление, а не цис или транс ; эта терминология отражает сложность использования исследований eQTL для анализа явного молекулярного механизма в этих особых случаи. Возможно, что более тонко, степень неравновесия по сцеплению также будет определять точность, с которой местоположение может быть определенным; в генетических панелях, созданных путем скрещивания с небольшим количеством рекомбинаций, или у близкородственных особей, длина генетически идентичной ДНК велика и может препятствовать разделению eQTL trans и его родственного гена, расположенного в одной и той же геномной области, даже на довольно больших расстояниях.
Таблица 2 показывает, что всего от 0,8% до 94% генов имеют eQTL в цис , и, как ожидается, наблюдаемое число зависит как от размера окна цис (геномное расстояние между и eQTL), используемые для определения связи/ассоциации cis , и на уровне значимости, используемом для определения связи/ассоциации между признаком и eQTL. Место (а) Регуляторные элементы недостаточно хорошо определены для большинства генов, и, следовательно, выбор размеров окна цис варьируется в исследованиях картирования eQTL, варьируясь от 10 т.п.н. в дрожжах (Brem et al.2002) до 20 Мб у мышей (Быстрых и др., 2005). Мы отмечаем, что есть задокументированные цис--действующие регуляторные элементы, которые расположены >1 Мб от гена, который они контролируют (Pfeifer et al. 1999), так что эти окна не являются неправдоподобными. Неудивительно, что чем больше цис -окно, тем больше вероятность найти признак экспрессии, расположенный в цис -окне соответствующего eQTL. Например, в наших исследованиях BXD с тремя тканями с использованием окна cis размером 5 Мб, ~2.6% связей находятся в -цис-, и эта доля увеличивается до ~8,0% при 20 Мб; в аналогичном исследовании на мышах Bystrykh et al. (2005) обнаружили 13,3% связей в цис , используя окно размером 20 Мб.
Хабнер и др. (2005), Monks et al. (2004) и Schadt et al. (2003) все сообщают, что доля цис--действующих eQTL увеличивается с более высокими порогами значимости сцепления или ассоциации, предполагая, что цис--действующие по сравнению с транс--действующие eQTL оказывают более сильное и более заметное влияние на экспрессию генов. .Петретто и др. (2006) проанализировали четыре ткани крысы и показали, что транс -eQTL обычно имеют меньший размер эффекта и более высокий уровень ложных открытий, чем цис -eQTL. В целом, общий вывод из этих исследований заключается в том, что в настоящее время предполагаемая цис -регуляторная вариация выявляется легче, чем транс .
Основные регуляторы экспрессии генов
Генетическая вариация в белке, таком как фактор транскрипции, который контролирует экспрессию нескольких генов, вероятно, воздействовать на все или большинство генов-мишеней.Этот класс изменчивости представляет особый биологический интерес, потому что, в принципе, его можно использовать для идентификации групп генов, имеющих общий контроль (формально «регулонов»). Коррелированная вариация в группах, а также отдельных генов, может иметь значительные фенотипические последствия.
Такие генетические вариации были обнаружены как eQTL, которые демонстрируют сцепление/ассоциацию с большим количеством генов, и они были названы главными регуляторами (Morley et al.2004). Правила определения «одного и того же» eQTL различны; некоторые исследования просто подсчитывают количество выражений, которые лучше всего сопоставляются с генетическим маркером независимо от того, является ли сцепление/ассоциация значимой на уровне всего генома или нет (Chesler et al. 2005), некоторые подсчитывают количество значимых связей всего генома на каждом маркере (Bystrykh et al. 2005; Hubner et al., 2005; Cotsapas, 2007), а некоторые сначала делят геном на ячейки, а затем подсчитывают количество значимых связей в каждой ячейке (Brem et al.2002 г.; Шадт и др. 2003 г.; Иверт и др. 2003 г.; Морли и др. 2004). В этих исследованиях было показано, что от 50% до 55% всех картируемых признаков экспрессии картируют до 17 главных регуляторов. (Таблица 2).
В нескольких сообщениях предполагается, что главные регуляторы могут быть вызваны систематическими артефактами микрочипов (Alberts et al. 2005; Li and Burmeister 2005; Williams et al. 2006). Реакции гибридизации микрочипов представляют собой сложные экспериментальные процессы, в которых измеряется комбинация биологического сигнала. и экспериментальный шум.Нормализация применяется к данным микрочипа, чтобы удалить любые источники небиологической изменчивости, такие как как различия в эффективности гибридизации внутри массива, но может быть трудно отделить биологический сигнал от шум. Обычно даже после нормализации на некоторых массивах группы генов могут иметь более высокую или более низкую экспрессию относительно остальные массивы. В контексте экспрессионной генетики группы генов, которые координированно изменяются в группе людей. может показать доказательства связи с одним eQTL, что приведет к появлению ложных главных регуляторов.
Несмотря на эти систематические артефакты, Monks et al. (2004) не удалось идентифицировать eQTL в линиях В-клеток человека, в которых количество картированных признаков экспрессии превышало случайное ожидание, предполагая, что главные регуляторы не являются универсальной функцией.
Множественные генетические регуляторы экспрессии генов
Как мы обсуждали выше, регуляция уровня мРНК одного гена, вероятно, зависит от многих факторов (см.1), и, таким образом, потенциально на него могут влиять более чем один генетический вариант. В традиционной генетической связи и полногеномной ассоциации исследования, в которых рассматривается один или небольшое количество количественных признаков, идентификация множественных генетических вариантов уже сложно из-за статистической сложности и больших вычислительных требований, предъявляемых комбинаторными вычислениями. требуется для проверки нескольких ассоциаций. Эта сложность усугубляется исследованиями картирования eQTL, в которых участвуют до десятков тысяч экспрессионных признаков представляют интерес, и поэтому почти невозможно выполнить систематический поиск множественных генетических влияний.По этой причине во всех анализах картирования eQTL, перечисленных в таблице 1, использовались методы с одним локусом, где в любой момент времени оценивается только один локус на предмет сцепления/ассоциации с каждым признаком экспрессии.
Хотя эти анализы явно не предназначены для обнаружения множественных влияний, признаки экспрессии, которые сопоставляются с более чем один eQTL действительно был идентифицирован (Brem et al. 2002; Schadt et al. 2003; Monks et al.2004 г.; Морли и др. 2004 г.; Брем и Кругляк 2005; Ченг и др. 2005 г.; Хабнер и др. 2005 г.; Стрейнджер и др. 2005 г.; Котсапас, 2007 г.).
Идентификация множественных пиков сцепления не является простой задачей; симуляционные исследования в сочетании с эмпирическими данными были проведены, чтобы предсказать, вероятно ли влияние на экспрессию генов множественных генетических локусов, и если да, то оценить количество локусов, которые могут способствовать изменению экспрессии генов (Brem et al.2002 г.; Брем и Кругляк, 2005). Эти результаты показывают, что только 3% признаков с высокой степенью наследуемости будут зависеть от одного локуса, в то время как >50% признаков вероятно, находятся под влиянием более чем пяти eQTL. Стори и др. (2005) представили метод мультилокусного картирования, разработанный для большого количества признаков экспрессии, и с помощью этого метода Brem et al. (2005) продемонстрировали, что на уровни мРНК 65% генов, находящихся под генетическим влиянием, влияют пары eQTL (пары локусов), в то время как только 13% пар локусов, идентифицированных с помощью метода картирования одного локуса, вероятно, будут иметь значительное взаимодействие.
Резюме
Анализ картирования eQTLможно использовать для анализа генетической регуляторной архитектуры экспрессии генов. Из исследований, которые были опубликованы, мы узнали, что: (1) генетические влияния на стационарные уровни мРНК являются обычным явлением; (2) цис--действующие варианты обнаруживаются легче, чем транс--действующие варианты; (3) на большинство транскриптов, находящихся под генетическим влиянием, вероятно, влияют множественные генетические варианты, и, таким образом, нам необходимо разработать дальнейшую методологию, которая может выполнять многолокусное картирование в этих условиях.В сочетании с прессованием необходимо увеличить размер картографических панелей для увеличения статистической мощности, это представляет собой огромную техническую проблему. Еще более разочаровывающим общим наблюдением является то, что возможность совмещать независимые исследования, даже проведенные на один и тот же организм, сильно скомпрометирован множеством картографических панелей, генетических маркеров, статистических методологий, гены на массивах и платформах массивов.
Биологическая интерпретация данных экспрессионной генетики
Техническая сложность генетического анализа экспрессии привела к разработке дополнительных аналитических методов в попытка использовать более полезную и потенциально непосредственно интерпретируемую биологическую информацию из данных.Эти исследования можно разделить на пять основных классов: (1) исследования, которые коррелируют вариации экспрессии с вариациями физиологических черты; (2) исследования, посвященные генам-регуляторам в eQTL; (3) исследования, посвященные регулируемым генам; (4) исследования, посвященные о генетических детерминантах уровней мРНК в известных сигнальных и метаболических путях; (5) тканевая специфичность генетических влияет.
Корреляция с физиологическими признаками
Рассмотрение уровней мРНК как «промежуточного фенотипа», который может связывать вариации генетической информации с физиологическими признаками. был основной движущей силой развития экспрессионной генетики, и Schadt et al.(2003), Быстрых и др. (2005), Чеслер и соавт. (2005), Hubner et al. (2005) и Li et al. (2006a) определили корреляцию между генетически обусловленной экспрессией и множеством признаков. Логика этих анализов такова. что ген с вариабельным уровнем мРНК, расположенный под пиком сцепления, связанным с физиологическим QTL, является потенциальным причинный кандидат. Конечно, если само изменение уровня мРНК напрямую коррелирует с физиологическим признаком, то это будет сильным подтверждением причинно-следственной связи.Быстрых и др. (2005) обнаружили восемь генов с цис--действующим генетическим влиянием, перекрывающимся с физиологическим QTL, влияющим на оборот гемопоэтических стволовых клеток (HSC), идентифицированных независимо друг от друга. де Хаан и др. (2002) у мышей BXD. Три гена были идентифицированы как гены-кандидаты в первоначальном исследовании, и все восемь содержали последовательность полиморфизмы. Чеслер и др. (2005) сопоставили уровни экспрессии гена Drd2 с опубликованными фенотипами из панели инбредных мышей BXD и выявили значимые корреляции для ряда фенотипов. включая предпочтение этанола и двигательную активность.
Идентификация генов-кандидатов-регуляторов в eQTL
Размер пиков сцепления eQTL означает, что существует несколько отчетов, в которых указывается причинная вариация в eQTL: Например, eQTL, определенные в штаммах RI, обычно содержат от 10 до 100 генов. Исключениями являются работы, о которых сообщает Zhu et al. др.(2004), Шадт и соавт. (2005) и Mehrabian et al. (2005), которые объединили данные о последовательности с картированием экспрессии и фенотипическими измерениями в сложном анализе с использованием условного моделирование независимости, которое выводит причинно-следственные связи между уровнями экспрессии мРНК и фенотипами. Эти подходы позволили Мехрабян и др. (2005), например, для идентификации 5-липоксигеназы в качестве гена предрасположенности к ожирению и костным признакам в eQTL ранее идентифицированный Schadt et al.(2003 г.) в гибриде F 2 C57BL/6J и DBA/2J.
Идентификация и проверка регулируемых генов или регулонов
Другие исследования пытались сосредоточиться на группах генов («регулонах»), на которые может влиять генетическая изменчивость: ключевой концепцией здесь является определение генов, чья коррелирующая картина экспрессии в разных фонах предполагает наличие общих регуляторов.Лан и др. (2006) дополнительно продемонстрировали полезность этого подхода на данных экспрессии из печени мышиной панели F 2 , разделяющей на ожирение и диабет; первоначально они идентифицировали гены, уровни мРНК которых, вероятно, регулируются цис или транс , а затем рассматривали эти гены как «зародыши» и расширяли состав этих наборов, идентифицируя эти гены. наиболее тесно связаны с ними. Анализ с использованием Gene Ontology показал, что многие из наборов генов, определяемых «семенем», были функционально связаны и/или функционировали в одних и тех же путях, участвующих в метаболизме липидов.Используя сцепленный ген Scd1 , который, как известно, участвует в метаболизме липидов и чувствительности к инсулину, авторы идентифицировали и экспериментально продемонстрировали что ген 3110032G18Rik взаимно регулируется с известными липогенными генами в различных контекстах.
Аналогичный подход был использован Ghazalpour et al. (2006), которые создали сети на основе коррелированных уровней мРНК генов, экспрессируемых в печени группы самок мышей из F 2 скрещивание C3H/HeJ и C57BL/6J.Путем определения групп потенциально совместно регулируемых генов (или «модулей») с использованием корреляции, а затем, идентифицировав eQTL, многократно связанные с генами в модуле, они определили модули, которые, вероятно, находятся под генетическим влияние: подход, который они описывают как «модуль-QTL» или mQTL. Их анализ идентифицировал набор коэкспрессируемых генов, которые в совокупности продемонстрировали связь с четырьмя eQTL и объяснили 70% дисперсии массы тела в популяции F 2 .
Идентификация генов с коррелированной экспрессией в разных генетических фонах даже при отсутствии идентифицируемых eQTL потенциально может раскрыть более широкий спектр влияний, чем обнаруженный анализом сцепления/ассоциации, хотя и с некоторыми трудностями. при оценке общей надежности с помощью показателей ложноположительных и ложноотрицательных результатов (дальнейшее обсуждение см. в Lan et al. 2006). Ли и др.(2006) выделили два механистически различных генетических влияния регуляторов, опосредованных изменениями численности ( транс -регуляторы экспрессии) и опосредованных структурными вариантами ( транс — регуляторы генотипа). Чтобы развить эти концепции, Lee et al. (2006) разработали и использовали Geronemo , который вычисляет вероятность, основанную на корреляциях, того, что изменчивость любой группы генов может быть объяснена комбинациями регуляторов экспрессии транс и транс генотипа; к ним относятся сигнальные молекулы, факторы, модифицирующие хроматин, факторы транскрипции и белки. участвует в процессинге, экспорте и посттранскрипционной модификации мРНК.Применение этого метода было продемонстрировано в 112 потомках дикого и лабораторного штамма Saccharomyces cerevisiae (данные Brem and Kruglyak 2005) и подчеркнули вероятную роль факторов ремоделирования хроматина, особенно комплекса SWI/SNF, в объяснении значительного доля наблюдаемой дисперсии экспрессии. Хотя методы, разработанные Lee et al. (2006), в принципе, применимы к регуляции высших эукариот, подход в настоящее время ограничен отсутствием конкретных функциональная аннотация многих генов у высших организмов.
На рисунке 2 мы выделяем другой возможный подход к раскрытию предполагаемой регуляторной архитектуры с использованием данных экспрессионной генетики из мозг, печень и почки на панели мышей BXD (Cotsapas 2007). Использование статистического подхода, называемого моделированием разреженного латентного фактора, для выявления группы из 24 генов, демонстрирующих доказательства. для корегуляции через коррелированные изменения уровней мРНК в мозге, печени и почках 31 мыши RI; генетическое картирование идентифицировали транс -eQTL на хромосоме (chr) 1 в головном мозге и chr 8 в печени: интересно, анализ с использованием базы данных связывания факторов транскрипции (JASPAR) продемонстрировали, что 11 из 24 генов были обогащены мотивом IRF2, а ген Irf2 расположен в eQTL chr 8 и экспрессировался во всех трех тканях (M.Коули, К. Котсапас, Р. Уильямс, Э. Чан, Дж. Пулверс, М. Лю, О. Луо, Д. Нотт и П. Литтл, в процессе подготовки).
Фигура 2.Регулоновый анализ генов. После анализа разреженного латентного фактора каждый ген представлен фиолетовой вершиной, соединенной с другими генами серой линией, если апостериорная вероятность корреляции между тремя тканями ≥0.90. Связь рисуется на хромосомах ниже (1–19, X, слева до справа ), если P < 10 −4 : цвет линии указывает на соответствующую ткань (синий, мозг, зеленый, почки, красный, печень ). Обратите внимание на группу генов справа от (обведены кружком), на которую влияет хромосома 1 в головном мозге и хромосома 8 в печени. В этом кластере генов происходит обогащение для мотива связывания Irf2; который также расположен в области сцепления на chr 8; и экспрессируется во всех трех тканях.
Исследования известных путей и взаимодействий генов с окружающей средой
Последняя группа исследований пытается изучить генетическое влияние на транскрипцию генов в известных метаболических или сигнальных пути.Хотя эти исследования по необходимости сосредотачиваются на относительно небольших наборах генов, для которых известно участие в метаболических путях, у них есть явное преимущество, заключающееся в том, что лежащий в основе клеточный механизм, по крайней мере, четко идентифицируется, что является значительным преимуществом. при рассмотрении последующей функциональной проверки. Используя этот подход, Ghazalpour et al. (2005) изучили 4670 генов в печени мышей в результате скрещивания F 2 BXD, о котором ранее сообщалось в Schadt et al.(2003), проанализировав 378 наборов функционально родственных генов, определенных KEGG, Biocarta и другими источниками аннотаций с использованием ранней версия процедуры Анализа обогащения набора генов (GSEA) (Subramanian et al. 2005) для идентификации генов, которые по-разному регулируются в любом заданном наборе. Они идентифицировали 170 генов, содержащихся в 13 путях. из которых девять связаны с циклом трикарбоновых кислот и три с метаболизмом холестерина.
Один из привлекательных аспектов этого подхода с точки зрения понимания функциональных взаимоотношений генотип-фенотип, это возможность прямого манипулирования фенотипами, например, путем прямой активации или ингибирования биохимического пути и изучение влияния генетической изменчивости на реакцию (например,г., Монтут и др. 2003 г.; Пассадор-Гургель и др. 2007). Ли и др. (2006b) исследовали взаимодействие eQTL и изменения температуры в RI-скрещивании двух штаммов Caenorhabditis elegans : интересно, что почти 60% генов, регулируемых транс-, демонстрировали признаки взаимодействия eQTL с температурой, но только 8% генов продемонстрировали цис -регуляцию, указывая на то, что контроль пути в этом случае преимущественно контролируется посредством транс взаимодействий.
Тканевая специфичность генетического влияния
В настоящее время степень, в которой генетическое влияние на уровни мРНК зависит или не зависит от ткани, остается неясной (Chesler et al. 2005; Cotsapas et al. 2006; Petretto et al. 2006; Yang et al. 2006). Данные микрочипов в областях мозга разных линий инбредных мышей ясно указывают на возможность специфичных для региона генетическое влияние экспрессии (Nadler et al.2006 г.; Ховатта и др. 2007). Наш собственный анализ изменчивости экспрессии у инбредных линий мышей подтверждает идею о том, что влияние генетической изменчивости на мРНК является по существу тканеспецифичным (Cotsapas 2007; (M. Cowley, C. Cotsapas, R. Williams, E. Chan, J. Pulvers, M. Liu, O. Luo, D. Nott, and P. Little, in prep. .), и с помощью на мышиной панели RI мы провели анализ картирования eQTL с использованием мРНК, выделенных из трех тканей: мозга, почек и печени. Из из 6075 транскриптов, которые экспрессируются во всех трех тканях, только два картированы с одним и тем же eQTL во всех трех тканях.То степень, в которой генетические влияния действительно тканеспецифичны, может иметь важное значение для исследований на людях, где доступ к разным тканям одного и того же человека обязательно ограничен.
Выводы
Возможно, самое поразительное наблюдение, которое мы можем сделать, подведя итог этому обзору, — это степень двусмысленности в позиционировании результатов экспрессионной генетики в биологическом контексте.Выше мы утверждали, что интерпретация данных сосредоточены на пяти областях; выявление прямых фенотипических последствий вариаций, изучение генов-регуляторов в eQTL, исследования были сосредоточены на регулируемых генах, и, наконец, исследования были сосредоточены на известных путях и тканях. Ясно, что это логично разработки любого генетического анализа; вопрос, который не затрагивается, заключается в том, в какой степени генетический анализ нет, соответствующий метод анализа.Отчасти ответ зависит от того, какая из областей рассматривается. Например, Можно ли легко интегрировать исследования полиморфизмов промоторов в проект ENCODE (Консорциум проекта ENCODE, 2007 г.)? Если да, нужна ли нам явная информация о сцеплении/ассоциации из экспрессионного генетического анализа или включение общих гаплотипов последовательностей ДНК в соответствующих экспериментальных анализах достаточно для определения цис- регуляторных вариантов? В целом, функциональные последствия вариаций последовательности одного гена можно эффективно изучать. и чувствительно с помощью подходов, основанных на молекулярной биологии, таких как трансгенез и анализ связывания промоторного белка / ДНК.Этим Аргумент, идентифицирующий цис -действующие влияния, возможно, является наименее привлекательным с экспериментальной точки зрения применением экспрессионной генетики. Напротив, -транс--действующие регуляторы, которые могут влиять на несколько генов, гораздо труднее идентифицировать с помощью молекулярно-биологических подходов. Поэтому использование генетических подходов привлекательно, поскольку, хотя их технически сложнее обнаружить, генетический анализ может привести к идентификации регулонов генов, находящихся под влиянием одних и тех же транс -действующих регуляторов.Описание регуляторной архитектуры, контролирующей экспрессию генов в больших масштабах, открывается существенное новое понимание, и будет большой проблемой интерпретировать фенотипические последствия, особенно в отношении для здоровья человека такого координированного изменения уровней мРНК.
Приблизились ли мы к пониманию того, как этот класс вариаций влияет на наше понимание эволюционных процессов? К сожалению, ответ должен быть однозначно отрицательным, и это отчасти потому, что у нас все еще есть чрезвычайно ограниченные экспериментальные данные о приравнивается ли изменение уровня мРНК к изменению уровня белка, и имеет ли это, в свою очередь, фенотипические последствия.То протеомная литература в этой области не сильна; у дрожжей Ghaemmaghami et al. (2003) обнаружили сильную корреляцию между уровнями белка и мРНК, а Lu et al. (2007) показывают, что 73% различий в уровнях белка объясняются обилием мРНК. Нам неизвестны подобные исследования на людях, но для белков плазмы Андерсон и Андерсон (2002) на основе обзора опубликованных исследований близнецов пришли к выводу, что в среднем 62% количественных вариаций уровней специфических уровней белка плазмы между людьми является генетическим по происхождению.Очевидная проблема заключается в том, как создать поле протеомной генетики?
Примечание добавлено в корректуру
Мы с восторгом читаем новую статью «Генетическая основа изменчивости протеома у дрожжей» (Foss et al. 2007), которая является первым исследованием, применяющим методологию экспрессионной генетики к генетическим влияниям на уровень белка — безусловно, Шаг в правильном направлении.
Благодарности
Мы благодарим нынешних и бывших коллег Криса Котсапаса, Дэвида Нотта, Марка Уилкинса, Флориана Брайтвейзера, Джунхонга (Оскара) Луо, Майклу Лю и Джереми Пулверсу за их вклад и понимание. Эта работа была поддержана грантом ARC Discovery Grant. (П.FRL), стипендия NHMRC Peter Doherty Fellowship (RBHW), австралийские награды для аспирантов (EKFC и MJC) и грант в помощь из Австралийского центра передовых вычислений и коммуникаций (PFRL).
Сноски
-
↵4 Текущие адреса: Школа медицинских исследований Джона Кертина, Австралийский национальный университет, Канберра, ACT 2601, Австралия;
-
↵5 CSIRO Livestock Industries, Queensland Bioscience Precince, 306 Carmody Road, St.Люсия, QLD 4067, Австралия.
-
↵6 Автор, ответственный за переписку.
↵6 Электронная почта p.little{at}unsw.edu.au; факс 61-2-9385-1483.
-
Статья размещена на сайте http://www.genome.org/cgi/doi/10.1101/gr.6981507
- Copyright © 2007, Cold Spring Harbour Laboratory Press
границ | Анализ генетической изменчивости и потенциальное применение в метаболическом моделировании в масштабе генома
1. Введение
Генная инженерия использовалась в течение нескольких десятилетий для манипулирования микроорганизмами с целью обеспечения производства ценных продуктов, включая первичные метаболиты (например, первичные метаболиты).например, аминокислоты и органические кислоты), вторичные метаболиты (например, антибиотики) и ферменты или другие рекомбинантные белки (Adrio and Demain, 2010). Таким образом, генная инженерия является центральной частью стремления к созданию устойчивых и эффективных процессов производства топлива, химикатов, пищевых ингредиентов и фармацевтических продуктов.
Большинство этих достижений были бы невозможны без технологий секвенирования, которые позволили нам идентифицировать генетические последовательности и подтвердить генетические манипуляции в микроорганизмах.Совсем недавно технологии секвенирования следующего поколения (NGS) предоставили нам возможность быстрого и дешевого секвенирования ДНК в беспрецедентных масштабах. NGS позволил собрать de novo геномов тысяч организмов, для которых ранее не было доступных геномных последовательностей, начиная от сложных многоклеточных организмов (Li et al., 2010; Nakamura et al., 2013; Pegadaraju et al., 2013). ; Kelley et al., 2014) к микроорганизмам (Soares-Castro and Santos, 2013; Yamamoto et al., 2014). Технологии NGS также предоставляют нам средства для повторного секвенирования организмов (Atsumi et al., 2010; Wang et al., 2014), то есть секвенирования генетически различных штаммов, которые достаточно близки к эталонному штамму с секвенированным геномом. Повторное секвенирование используется для определения генетических вариантов, начиная от однонуклеотидных вариантов (SNV) и заканчивая более сложными структурными вариантами, такими как большие делеции, инверсии и транслокации. Падение стоимости секвенирования позволяет проводить рутинное повторное секвенирование штаммов, выделенных из дикой природы, контролировать генетическую стабильность производственных штаммов во время процессов генной инженерии и ферментации, а также определять генетическую основу адаптивной лабораторной эволюции (ALE) (Herrgård and Panagiotou, 2012). .В дополнение к биотехнологическим применениям повторное определение последовательности микробных штаммов также играет ключевую роль в других областях, таких как эпидемиология инфекционных заболеваний, вызываемых бактериальными и грибковыми патогенами, а также в понимании влияния деятельности человека на микробное разнообразие и эволюцию в окружающей среде.
Метаболические модели в масштабе генома (GSM), состоящие из биохимических реакций и их связи с геномом и протеомом клетки [через ассоциации ген-белок-реакция (GPR)], являются проверенной основой для анализа in silico Метаболическая физиология микробов.Метаболические модели в масштабе генома также успешно использовались для конструирования метаболически сконструированных штаммов с улучшенной продукцией коммерчески ценных белков и метаболитов: рекомбинантных антител, пищевых добавок (например, ванилина), органических кислот, этанола и др. (Теппер и Шломи, 2009; Брочадо и др., 2010). Эти модели становятся все более популярными в последнее десятилетие, и на сегодняшний день опубликовано более 100 моделей для различных организмов (http://optflux.орг/модели). Самая большая сила GSM заключается в их простоте и вычислительной эффективности; новые GSM могут быть легко построены из геномных аннотаций, дополненных ограниченными экспериментальными данными, а прогнозы из GSM могут быть получены с использованием стандартных методов математической оптимизации (Varma and Palsson, 1993; Segre et al., 2002; Shlomi et al., 2005), позволяющих фенотипически прогнозы в течение нескольких минут.
Генетическая изменчивость, которая влечет за собой полную потерю функции, обычно называемая нокаутом гена, успешно использовалась для адаптации GSM к определенному генотипу для улучшения производства ценных соединений [e.g., биобутанол (Lee et al., 2008), сесквитерпен (Asadollahi et al., 2009), ванилин (Brochado et al., 2010), полигидроксиалканоаты (Puchalka et al., 2008) или L-валин (Park et al., 2010). al., 2007)], но до сих пор не разработана методологическая база, которая позволила бы систематически включать другие типы генетических вариантов. В этой работе мы рассматриваем существующие инструменты для анализа генетических вариантов, которые фиксируют более тонкие изменения, такие как синонимичные и несинонимичные SNV в кодирующих областях или варианты в промоторных или других регуляторных областях.Мы сосредоточимся на описании проблем, связанных с объединением более тонкой информации о генетических вариантах с GSM, чтобы использовать модели для прогнозирования специфических для штаммов фенотипов.
2. Выявление последствий генетической изменчивости
2.1. Генетическая изменчивость
Генетические варианты, включая SNV и более крупные структурные варианты, обычно наблюдаются при повторном секвенировании природных или сконструированных штаммов (рис. 1). SNV можно найти в геноме в различных функциональных областях: (i) последовательности, кодирующие белок, (ii) промоторы и другие регуляторные элементы, такие как сайты связывания рибосом, (iii) сайты сплайсинга и другие области, влияющие на структуры транскриптов, и (iv) другие геномные области с неизвестными прямыми связями с любой данной функцией белка.Более того, вставки или делеции нуклеотидов (вставки) в кодирующей области могут вызывать сдвиг в открытой рамке считывания, обычно обозначаемый как мутации со сдвигом рамки (рис. 1А). На уровне структуры генома хромосомные перестройки, например, замены, инверсии, делеции и вставки, могут влиять на функцию одного или нескольких белков (рис. 1В).
Рисунок 1. Распространенные генетические вариации . Вариации на уровне нуклеотидов (A) и структурном уровне (B) . (C) Однонуклеотидный полиморфизм A/T в популяции.
Спектр результирующих эффектов, вызванных этими генетическими вариациями, на функцию или экспрессию отдельных генов или белков, очень широк. Несинонимичные SNV или индели внутри рамки считывания в последовательностях, кодирующих белок, могут нарушать, усиливать или модифицировать активность белка в зависимости от внесенной точной аминокислотной замены. Ожидается, что введение или удаление стоп-кодона с помощью специфических SNV или внерамочных вставок приведет к более радикальным изменениям функции белка.Например, появление стоп-кодона может привести к разделению многодоменного белка на несколько отдельных однодоменных белков. Удаление или замена стоп-кодона может вызвать трансляционное прочтение, ведущее к удлиненному белку с потенциальными новыми функциями (Long et al., 2003). SNV и вставки в регуляторных областях, таких как промоторы, могут влиять на процессы транскрипции или трансляции, вызывая изменение уровней экспрессии в определенных белках. У эукариот варианты внутри интронов также могут влиять на структуры транскриптов, вводя новые экзоны или удаляя существующие.Некоторые вариации также могут быть полностью бесшумными без изменения фенотипа, например, изменение положения стоп-кодона может не изменить активность белка. В идеале мы должны быть в состоянии предсказать степень, в которой одиночные и множественные генетические варианты внутри или рядом с кодирующим локусом влияют на соответствующую функцию или экспрессию белка. Это позволило бы нам быстро разобраться в огромном количестве данных повторного секвенирования, которые становятся доступными, без необходимости экспериментально проверять эффекты всех вариантов.
Крупномасштабные структурные вариации, такие как дупликации, делеции, транслокации и инверсии, могут оказывать существенное влияние на экспрессию или активность отдельных белков. Например, может иметь место полная потеря одного или нескольких генов, или дупликация геномных областей может изменить экспрессию нескольких генов внутри или рядом с этими областями (Blount et al., 2012). Очень крупномасштабные геномные изменения, такие как удвоение полных хромосом, могут одновременно изменить активность сотен белков, о чем сообщалось как у природных микробных штаммов (Gordon et al., 2009) и в штаммах, созданных методом ALE (Caspeta et al., 2014). Эффекты структурной геномной изменчивости часто более системны, чем эффекты вариаций меньшего масштаба, но любая структура, пытающаяся предсказать фенотипические эффекты генетической изменчивости, должна учитывать как мелкомасштабные, так и крупномасштабные вариации.
2.2.
In silico : Прогнозирование влияния генетических вариантовОсновная проблема понимания фенотипических последствий генетической изменчивости заключается в нашей способности предсказывать механистические последствия мутаций.Белки представляют собой очень сложные структуры, которые попадают в различные функциональные категории и могут характеризоваться многими различными свойствами. Например, то, как измеряется активность белков, зависит от их функциональной категории: факторы транскрипции можно охарактеризовать по их силе связывания с определенной промоторной областью, в то время как метаболические ферменты обычно характеризуются их каталитической активностью и специфичностью в отношении определенного субстрата. Более того, белки не действуют изолированно, а взаимодействуют друг с другом и с метаболитами, и эти взаимодействия влияют на активность белков.Здесь мы предоставляем неисчерпывающий обзор типов методов, которые обычно используются для прогнозирования влияния генетических вариантов на функцию белка.
Изучение однонуклеотидных полиморфизмов (SNP), влияющих на здоровье человека, является одним из основных направлений современных медицинских исследований. В генетике человека SNP представляют собой замены одиночных нуклеотидов, встречающиеся более чем у 1% населения. Было реализовано несколько алгоритмов для определения влияния SNP, в основном специализированных для анализа данных генотипирования человека (см. Таблицу 1 и Рисунок 2).Одним из ограничений большинства этих алгоритмов является то, что они представляют собой бинарные классификаторы — вредные или нейтральные, вызывающие болезни или нейтральные, терпимые или нетерпимые. Это означает, что предполагается, что генетические изменения либо не будут иметь никакого эффекта, либо окажут некоторое измеримое негативное влияние на фенотип. Это не может быть проблемой в контексте заболеваний человека, поскольку данные SNP в основном используются в диагностике. Однако для точной настройки сконструированных микробных штаммов требуется нечто большее, чем черно-белый подход для прогнозирования различных эффектов на функцию белка.Это связано с тем, что многие генетические варианты могут давать белки как с повышенной, так и с пониженной активностью, что требует методов, способных также предсказать потенциальное усиление или модификацию функций. В частности, когда применяются методы мутагенеза и селекции или ALE, обычно наблюдается усиление функциональных мутаций специфических генов, которые имеют решающее значение для адаптации, например, к новым источникам углерода (Conrad et al., 2011).
Таблица 1 . Обзор доступных программных инструментов для прогнозирования влияния генетических вариантов .
Рисунок 2. Сводка свойств и подходов к программному обеспечению, указанному в таблице 1 . Найденные подходы делятся на четыре разные категории: Машинное обучение , Вероятностный , Оценка (вычисление суммирующей оценки набора отобранных вручную статистических данных) и Правило (с использованием набора эмпирически полученных правил). .Каждый из этих подходов обеспечивает один из двух типов классификаций: бинарную классификацию (например, нейтральную или вредную) или множественную классификацию (например, доброкачественную, нейтральную и вредную). Характеристики, используемые этими подходами, могут быть вычислены на основе свойств следующих пяти категорий: (i) физико-химические свойства (например, доступность растворителя, полярность, заряд, беспорядок и Грэнтэм), (ii) структурная информация о первичных, вторичных, и третичная структура белка (например,, α-спирали, β-листы и спираль), (iii) эволюционные свойства (множественное выравнивание последовательностей, позиционно-специфические оценочные матрицы и скрытые марковские модели) и (iv) аннотация генома (термины GO или другие аннотации функций белка) . Поддерживаемые варианты определялись либо путем доступа к веб-сайтам инструментов, либо по описанию самого подхода.
Из существующих алгоритмов (таблица 1) SIFT ( S orting I ntolerant f rom T olerant) (Ng and Henikoff, 2001) часто используется в качестве золотого стандарта для сравнения производительности алгоритмов. новых алгоритмов или в качестве основы для новых стратегий прогнозирования.SIFT и родственные подходы основаны на представлении о том, что эволюционную консервацию можно использовать для предсказания функциональной важности каждой аминокислоты в белке и влияния конкретных аминокислотных замен. В этих методах обычно используется множественное выравнивание последовательностей родственных белков для определения вероятностного описания того, какие аминокислотные замены разрешены в определенных участках целевого белка. Эти описания можно использовать для определения вероятности того, что несинонимичные кодирующие SNP, наблюдаемые в наборе данных повторного секвенирования, будут допустимы белком; замены с оценкой вероятности ниже порогового значения считаются вредными (Kumar et al., 2009).
Сортировка непереносимых от переносимых обеспечивает только бинарную классификацию вредных/невредных, и были разработаны другие методы, позволяющие прогнозировать случаи, когда SNP улучшают функцию белка. Подходы Polyphen (Ramensky, 2002) и PolyPhen2 (Adzhubei et al., 2010) позволяют различать три состояния при анализе действия SNP: доброкачественное, нейтральное или вредное. Polyphen использует список предопределенных правил, которые объединяют выходные данные нескольких алгоритмов с использованием комбинаций структурных и основанных на последовательностях показателей воздействия мутаций. PolyPhen2 использует подход машинного обучения (наивная байесовская модель) для прогнозирования общей оценки эффекта варианта, а классификация по трем категориям основана на пороговых значениях. Хотя алгоритм обучается на наборах данных человека, аналогичные методы потенциально могут быть использованы для построения прогностических моделей различных эффектов у микроорганизмов. Общая оценка эффекта варианта также может быть использована в более продвинутых методах, которые объединяют оценки разных вариантов, влияющих на разные белки, для прогнозирования фенотипа.
Большинство исследований генетической изменчивости сосредоточено на SNP и игнорирует вставки, которые также обычно наблюдаются при сравнении родственных микробных штаммов друг с другом. Подходы PROVEAN (Choi et al., 2012) и Mutation tester 2 (Schwarz et al., 2014) позволяют анализировать как SNP, так и вставки. PROVEAN использует показатели матрицы замещения (т. е. BLOSUM62) со штрафами за пробел и расширение для вычисления показателя вариации между диким типом и мутантом.Совсем недавно Mutation tester 2 вычисляет несколько характеристик (структурные и эволюционные свойства) для мутировавшей последовательности с использованием байесовского классификатора.
Одним из возможных подходов к улучшению нашей способности предсказывать влияние вариантов на функцию белка было бы предсказание эффектов изменений аминокислот на стабильность и фолдинг белка (Khan and Vihinen, 2010). Для этих задач доступен ряд инструментов (Khan and Vihinen, 2010), и прогнозы стабильности можно использовать для прогнозирования различных эффектов на функцию белка, поскольку сильные дестабилизирующие мутации приведут к полной потере функции белка.В независимых оценочных исследованиях было обнаружено, что методы прогнозирования влияния вариантов на стабильность белка являются умеренно точными (Khan and Vihinen, 2010). По этой причине предикторы стабильности следует комбинировать с другими подходами к предсказанию эффекта варианта, чтобы улучшить их прогностическую способность для общего анализа эффекта варианта. Применение этих типов методов прогнозирования стабильности будет обсуждаться более подробно в разделе 3.2 вместе с приложениями метаболического моделирования.
Большинство алгоритмов (53%) для прогнозирования эффекта варианта, перечисленных в таблице 1, основаны на подходах машинного обучения [например, AUTO-MUTE (Masso and Vaisman, 2010), FunSAV (Wang et al., 2012) или HANSA ( Acharya and Nagarajaram, 2011)], что является практической стратегией, учитывая огромное количество доступных данных о заболеваниях человека. Что касается выбора признаков, то большинство методов используют информацию об эволюционном сохранении (92%), и более половины полагаются на структурные свойства (69%). Выбор достаточных функций сам по себе является сложной задачей; независимо от того, какой подход используется, необходимо определить, какие свойства и атрибуты белков способны различать интересующие фенотипы.Улучшения в возможностях прогнозирования, обеспечиваемые функциями, основанными на последовательности, эволюции или структуре, изучались ранее, и эти исследования показали, что включение структурных свойств приводит к значительному улучшению прогнозирующей способности (Saunders and Baker, 2002). Это было недавно подтверждено эталонным тестом производительности, включающим несколько существующих алгоритмов (Thusberg et al., 2011). Еще одной попыткой сравнить и улучшить различные подходы является сообщество «Критическая оценка интерпретации генома» (CAGI), которое организует соревнование по оценке влияния генетических вариантов на известные фенотипы заболеваний.
В то время как большинство алгоритмов нацелены на предсказание различных эффектов на отдельные белки, другой цели преследует метод SNP-IN, который предсказывает, как SNP влияет на белок-белковые взаимодействия (PPI) (Zhao et al., 2014). Это достигается за счет набора признаков, который включает относительное изменение свободной энергии между ИПП дикого типа и мутантного типа, энергию всех взаимодействий в белковом комплексе и другие физико-химические свойства, например, гидрофобную сольватацию или водные мостики.Используя эти функции, контролируемые и полуконтролируемые подходы к машинному обучению используются для прогнозирования того, насколько вредны SNP. Этот подход очень интересен, поскольку изменения в PPI можно использовать для объяснения эпистатических взаимодействий между несколькими вариантами. Как и для некоторых ранее упомянутых алгоритмов прогнозирования, для SNP-PI требуется существующая трехмерная модель структуры белка и, кроме того, знание PPI, в которых участвует данный белок.
В более широком масштабе исследования ассоциаций всего генома используются для определения того, как различия между сотнями тысяч людей и приводят к последствиям от генотипа к фенотипу.Эти подходы работают как черные ящики и используют статистические подходы и подходы машинного обучения, требующие огромных наборов данных. Текущая работа и приложения (например, оценка клинического риска) были недавно проанализированы (Okser et al., 2014).
2.3.
In vivo : Глубокое мутационное сканирование и TN-SEQСеквенирование следующего поколения позволило изучить влияние генетической изменчивости на отдельные белки или регуляторные элементы in vivo и in vitro .Глубокое мутационное сканирование (DMS) является эффективным высокопроизводительным методом измерения влияния мутаций на стабильность и функцию белка (Fowler and Fields, 2014). Пространство всех возможных аминокислотных замен в белке тщательно проверяется путем сначала создания библиотеки вариантов последовательностей с использованием стандартных методов, таких как подверженная ошибкам ПЦР, а затем с помощью высокопроизводительного анализа для выбора вариантов на основе показателя пригодности (например, скорость роста, связывание лиганда или флуоресценция продукта) и, наконец, путем применения глубокого секвенирования к выбранным и невыбранным пулам вариантов последовательностей.Результатом этого подхода является матрица, содержащая значения приспособленности для каждой аминокислотной замены, обнаруженной в выбранном пуле. В зависимости от метода, используемого для создания разнообразия последовательностей и глубины секвенирования, DMS также можно использовать для измерения эпистатических эффектов между заменами в разных сайтах.
Применимость DMS в первую очередь ограничена отсутствием высокопроизводительных функциональных анализов для большинства белков, и до сих пор DMS не применялся к метаболическим ферментам. Когда DMS может применяться в более широком масштабе, результаты, полученные в результате анализа, могут повысить прогностическую способность биоинформационных инструментов для анализа генетической изменчивости за счет предоставления более полных наборов обучающих данных для типов прогностических методов, обсуждавшихся в предыдущем разделе.Методы, подобные DMS, также можно использовать для систематического изучения эффектов генетической изменчивости в регуляторных областях на экспрессию белка с использованием анализов на основе флуоресцентных белков.
Здесь мы выделим несколько тематических исследований с использованием DMS и родственных методов для изучения функции белка или регуляторного элемента. При анализе поли(А)-связывающего белка Saccharomyces cerevisiae (Melamed et al., 2013) были обнаружены сильные эпистатические эффекты между заменами в определенных сайтах.Хотя эпистаз не был широко распространен, это вызывает беспокойство с точки зрения компьютерного моделирования, поскольку подходы к моделированию обычно не учитывают эпистаз. Еще одним важным моментом является идентификация альтернативных стартовых кодонов. Хотя анализ в предыдущих исследованиях, DMS показал, что некоторые аминокислоты могут быть заменены метионином и давать функциональные белки (Kim et al., 2013). Эта биологическая информация может быть экстраполирована на другие исследования и имеет большое значение при разработке стратегий для понимания влияния мутаций in vivo или in silico .Стратегии, подобные DMS, также использовались для систематического изучения эффектов изменчивости сайтов связывания факторов транскрипции и других регуляторных элементов, таких как сайты связывания рибосом (Kosuri et al., 2013). Эти исследования заложат основу для прогнозирования влияния вариантов некодирующей последовательности на экспрессию белка.
Описанные выше методы позволяют нам систематически изучать эффекты большого количества вариантов в отдельных белках или регуляторных областях.В микроорганизмах также можно использовать основанный на секвенировании метод следующего поколения, называемый Tn-seq, для систематического изучения влияния разрушения большого количества геномных локусов на клеточные фенотипы (van Opijnen and Camilli, 2013). Транспозоны — это мобильные элементы ДНК, которые могут разрушать генетический локус, интегрируясь в него (рис. 1В). Tn-seq с использованием библиотек вставок транспозонов с высокой плотностью можно использовать для изучения функции, например, регуляторных элементов и специфических белковых доменов в едином полногеномном анализе (van Opijnen and Camilli, 2013).Tn-seq нашел множество применений в микробиологии и использовался для идентификации функции генов, понимания организации генома, картирования генетических взаимодействий или оценки эссенциальности генов (van Opijnen and Camilli, 2013; Yang et al., 2014). Tn-seq не дает разрешения на уровне одной пары оснований, но этот метод можно быстро использовать для получения информации на уровне субгенов, относящейся, например, к существенности определенных доменов в белке. Эта информация, в свою очередь, может быть использована для улучшения прогнозов эффектов вариантов, поскольку варианты в основных доменах белка с большей вероятностью будут предсказаны как вредные, чем варианты в несущественных доменах того же белка.
3. Прогнозирование фенотипов по генотипам в масштабе генома
3.1. Статистические и сетевые подходы к прогнозированию фенотипов по генотипам
Раздел 2 посвящен задаче прогнозирования эффектов генетической изменчивости на функцию или экспрессию отдельных белков. Однако это лишь малая часть гораздо более крупной проблемы, заключающейся в предсказании фенотипических эффектов клеток или организмов всех генетических вариантов, присутствующих в геноме. Это требует учета влияния вариаций на функцию и экспрессию всех белков.До сих пор было предпринято на удивление мало попыток взять все генетические варианты, обнаруженные у человека (человека или микробного штамма), и попытаться предсказать, как все эти варианты вместе взятые повлияют на определенные фенотипы (Burga and Lehner, 2013; Lehner , 2013).
Одной из первых систематических попыток достижения этой цели было новаторское исследование Jelier et al. в S. cerevisiae , где фенотипы роста выбранных штаммов дрожжей в различных условиях были предсказаны на основе генетических различий между эталонным штаммом и интересующим штаммом (Jelier et al., 2011). Это было достигнуто за счет первого предсказания эффектов кодирующих и регуляторных вариантов на функцию и экспрессию белка с использованием подходов, подобных описанному в предыдущем разделе. Затем эти предсказания эффекта варианта были объединены в одно предсказание фенотипа для штамма с использованием опубликованных данных фенотипирования роста с делецией одного гена для эталонного штамма дрожжей в тех же условиях. Этот подход можно считать очень упрощенным, поскольку эффекты нескольких генетических вариантов, действующих на отдельные белки, рассматривались как кумулятивные.Несмотря на это, подход по-прежнему позволял точно прогнозировать фенотипы роста в широком диапазоне условий. Также существует ряд других подходов для прогнозирования более широких фенотипических последствий отдельных вариантов путем сопоставления данных вариантов с биологическими сетями, такими как PPI или генетические сети (Carter et al., 2013). Однако в этих подходах обычно не пытались использовать весь генотип индивидуума (т. е. более одного варианта за раз) для предсказания конкретных фенотипов.
3.2. Использование метаболических моделей в масштабе генома для интерпретации генетических вариантов
Описанные выше методы прогнозирования фенотипа основаны на данных и используют статистические модели для прогнозирования эффектов генетических вариантов в контексте биологических сетей. Однако для метаболических сетей мы можем выйти за рамки статистических моделей и основанных на графах описаний и перейти к моделям, основанным на ограничениях, которые масштабируются до уровня генома и включают физико-химические ограничения, пропускную способность и направленность реакций [см. Price et al.(2004) для обзора моделирования на основе ограничений]. Этот тип механистического подхода к моделированию очень полезен для понимания генетических изменений, влияющих на определенные метаболические фенотипы. Например, изучение SNP, влияющих на митохондриальный метаболизм (Jamshidi and Palsson, 2006), является хорошим примером того, как данные о вариантах могут быть отображены на метаболических сетях для объяснения механистической основы фенотипов заболеваний.
Метаболические модели в масштабе генома состоят из биохимических реакций, собранных из литературы и аннотации генома организма.Эта система реакций закодирована в виде матрицы стехиометрических коэффициентов, которую обычно называют матрицей стехиометрии. Если предположить, что метаболизм находится в стационарном состоянии, т. Е. Концентрации метаболитов не меняются с течением времени, все потоки должны уравновешивать друг друга. Эти балансы потоков представляют собой линейные ограничения, которые можно легко проанализировать с помощью методов линейной алгебры.
Кроме того, после включения дополнительных ограничений, например, известных скоростей поглощения и секреции и знаний о направленности реакции, методы линейной оптимизации могут вычислять биологически значимые векторы потоков, которые максимизируют определенные целевые функции.Например, рост можно смоделировать путем максимального потребления предшественников биомассы в эмпирически определенных пропорциях. Этот тип анализа обычно называют анализом баланса потоков [FBA; см. Orth et al. (2010) для всестороннего введения в этот метод].
Глобальные оптимальные решения для этих задач линейной оптимизации могут быть очень эффективно рассчитаны с использованием линейного программирования (время вычислений составляет от миллисекунды до секунды для моделей в масштабе генома). Таким образом, можно вычислить тысячи фенотипов за несколько минут, просто изменив ограничения задачи [см. Lewis et al.(2012) для полного списка доступных методов in silico и (Bordbar et al., 2014) для обзора их приложений].
Поскольку отношения между реакциями, ферментами и генами (обычно называемые ассоциациями GPR) обычно известны и закодированы в этих моделях, эффект нокаута гена можно легко сопоставить с ассоциированными реакциями, ограничивая их потоки равными нулю или удалением из модели. Таким образом, FBA можно использовать для вычисления метаболического фенотипа, связанного с делецией метаболического гена, что делает его пригодным для анализа данных генетической изменчивости, включающих делеции или другие мутации, приводящие к полной потере функции ферментов.
Анализ баланса потоков предполагает, что нокаутные штаммы могут восстанавливаться до оптимального фенотипа роста, что может быть нереалистичным в случаях, когда механизмы регуляции, явно не смоделированные в этих моделях, могут быть не в состоянии обеспечить желаемое состояние. Другие методологии [например, ROOM (Shlomi et al., 2005), MoMA (Segrè et al., 2002), MiMBl (Brochado et al., 2012) и RELATCH (Kim and Reed, 2012)] используют более правдоподобные предположения и было показано, чтобы улучшить точность прогнозов нокаута.Например, MoMA минимизирует евклидово расстояние распределений потока дикого типа и мутанта, предполагая, что мутант достигает ближайшего возможного распределения потока, которое не обязательно является оптимальным. Предсказательная сила FBA и этих других подходов была тщательно оценена с использованием полногеномных анализов нокаута генов (Snitkin et al., 2008) и библиотек вставок транспозонов (Yang et al., 2014) и в целом привела к высокой степени точности. (Монк и Палссон, 2014).
Модели, основанные на ограничениях, также применялись для прогнозирования эпистатических взаимодействий путем имитации эффектов парных делеций генов, но со значительно меньшей точностью по сравнению с одиночными делециями (Szappanos et al., 2011). Кроме того, моделирование множественных делеций генов успешно применялось при разработке стратегий проектирования для метаболической инженерии путем перенаправления потока на желаемые продукты (Milne et al., 2009; Blazeck and Alper, 2010).
Ряд ограничивающих факторов может снизить способность моделей, основанных на ограничениях, предсказывать фенотипические эффекты мутаций с потерей функции: (i) отсутствующие реакции и ошибочные GPR, (ii) ошибочные ограничения потока из-за отсутствия термодинамической или регуляторной информации, и (iii) предположение о фиксированном составе биомассы, который, как известно, меняется в зависимости от условий роста.Даже с этими ограничениями модели, основанные на ограничениях, по-прежнему превосходят статистические модели в предсказании последствий делеций генов (Szappanos et al., 2011).
Поскольку модели, основанные на ограничениях, продемонстрировали хорошую способность предсказывать фенотипические результаты одиночных и множественных делеций генов, эти модели также должны быть полезны для предсказания эффектов других генетических вариантов. SNV или indel, которые, по прогнозам, уменьшают максимальную скорость потока фермента, могут использоваться для ограничения верхней границы потока.FBA и подобные методы могут быть использованы для вычисления эффектов этих вариаций на фенотип, обеспечивая общесистемный обзор эффектов, вызванных заменой (Jamshidi et al., 2007). Это быстрый и эффективный способ предсказания фенотипов, но он требует, чтобы можно было оценить влияние варианта на максимальную скорость переноса. Тем не менее, случаи полной потери функции попадают в ту же категорию, что и нокауты генов, и сочетающие в себе инструменты биоинформатического прогнозирования, обсуждаемые в разделе 2.2 с возможностями моделирования можно использовать для интеграции данных вариантов. Этот подход также можно распространить на любое количество вариантов и генов с оговоркой, что эпистатические взаимодействия в настоящее время не точно фиксируются моделями.
В настоящее время существует лишь ограниченное количество исследований, в которых используются GSM для систематического изучения влияния генетических вариантов на фенотипы. Чанг и др. (2013) провели исследование, в котором GSM в сочетании с белковыми структурами метаболических ферментов (GEM-PRO) использовались для интерпретации данных о генетических вариантах штаммов Escherichia coli , которые эволюционировали, чтобы переносить высокие температуры (Chang et al., 2013). В этом исследовании GSM E. coli был ограничен с использованием экспериментально или биоинформационно определенных термостабильностей метаболических ферментов. Поскольку максимальная пропускная способность реакции пропорциональна концентрации активного фермента, изменения температуры можно моделировать путем соответствующего изменения ограничений потока. Это позволяет прогнозировать ферментативные стадии, которые непропорционально чувствительны к температуре. Для эволюционировавших штаммов использовали анализ баланса потоков для изучения адаптации мутировавших ферментов; ограничения, связанные с мутировавшими белками, были ослаблены, чтобы объяснить экспериментально измеренные скорости роста (Chang et al., 2013). Исследование не включало отдельные прогнозы влияния вариантов на функцию белка, а рассматривало все варианты, наблюдаемые в белке, как потенциально влияющие на его активность.
В более позднем исследовании Nam et al. (2014) описывает использование GSM для понимания метаболических эффектов раковых мутаций. В частности, Nam et al. использовать информацию о генетических мутациях, данные профиля экспрессии генов и GSM человека (Thiele et al., 2013) для построения контекстно-зависимых моделей для различных типов рака.Потеря и приобретение функции были систематически проанализированы. Потерю функции моделировали, как описано выше (т. е. ограничивая потоки затронутых реакций до 0). Приобретение функции, с другой стороны, было смоделировано путем добавления новых беспорядочных действий, предсказанных хемоинформационными подходами. Этот подход позволил прогнозировать потенциальные онкометаболиты.
3.3. Кинетическое моделирование генетических вариантов
Как упоминалось в предыдущем разделе, моделирование на основе ограничений не предоставляет никакой информации о динамическом поведении метаболической системы.Полное кинетическое описание сети биохимических реакций можно сформулировать с помощью обыкновенных дифференциальных уравнений (Heinrich and Schuster, 1996). Основное преимущество использования кинетических моделей для изучения эффектов генетической изменчивости заключается в их способности учитывать мутации, влияющие на каталитические или регуляторные сайты фермента, вызывающие повышение или потерю каталитической активности или сайты связывания аллостерических регуляторов.
Предыдущие исследования метаболизма эритроцитов дают обзор того, как SNP могут изменять кинетические параметры и как кинетические модели могут использоваться для объяснения метаболических синдромов, вызванных дефицитом ферментов (Jamshidi, 2002; Jamshidi and Palsson, 2009).Недостатком использования кинетических моделей является то, что кинетические параметры недоступны для большинства ферментов, и их измерение может быть затруднительным. По этой причине создание прогнозирующих кинетических моделей в масштабе генома остается проблемой (Stanford et al., 2013). Кинетические модели являются жизнеспособным инструментом для интерпретации данных о генетических вариантах только в конкретных случаях, таких как, например, эритроцит с относительно простым метаболизмом.
4. Соображения и будущие направления
4.1. Методы и инструменты для прогнозирования влияния генетических вариантов
За последнее десятилетие было изучено множество подходов к пониманию и анализу эффектов генетической изменчивости. В частности, наиболее активной областью было применение методов NGS для характеристики генетической изменчивости в контексте болезней человека. Объем информации, связанной с заболеванием, делает подходы машинного обучения очень подходящими для прогнозирования эффектов отдельных генетических вариантов. Поскольку большинство методов прогнозирования были обучены и протестированы на данных человека, многие из существующих методов не работают так же хорошо или просто не подходят для анализа микробных генетических вариантов.
Другой областью, в которой изучение генетической изменчивости микробов отстает от генетики человека, является систематический сбор данных о вариантах и фенотипировании. В настоящее время предпринимаются усилия по стандартизированному сбору данных о генотипе и фенотипе человека с использованием таких баз данных, как dbSNP и Европейский архив вариаций. База данных UniProt также собирает варианты, обнаруженные в последовательностях белков, когда эта информация доступна. Каждый день во всем мире секвенируются тысячи новых экологических или патогенных изолятов и лабораторно разработанных микробных штаммов, но общедоступного централизованного хранилища этих данных не существует.Мы утверждаем, что чрезвычайно важно собирать данные о генетических вариантах вместе с соответствующими фенотипическими данными стандартным для микробов способом.
Все существующие алгоритмы предсказания эффекта варианта используются для классификации вариантов по заранее назначенным категориям (например, вредоносные или невредные). Подходы, которые предсказывают вредные эффекты, уже могут рассматриваться как нокауты при моделировании их фенотипических эффектов с использованием GSM, но этот подход упускает из виду более тонкие эффекты мутаций.Чтобы улучшить нашу способность предсказывать фенотипы, необходимо выйти за рамки классификации и перейти к количественным измерениям различных эффектов на функцию отдельных белков. Существует множество особенностей, связанных с функцией белка, которые могут иметь значение для предсказания вариантов эффектов: эволюционные и консервативные, физико-химические (например, заряд, полярность или свободная энергия) и структурные (например, вторичные структуры, пространственные расстояния между аминокислотами или B). -факторы).
Существующие методы прогнозирования вариантов эффектов в первую очередь сосредоточены на общих предикторах для всех белков, независимо от их функции (например,г., ферменты, факторы транскрипции, транспортеры, шапероны и др.) и как они ведут себя в окружающей среде (т. е. взаимодействуют с другими элементами: белками, метаболитами, ДНК и др.). Это ограничивает предсказательную силу методов в тех случаях, когда легкодоступна дополнительная информация, например относительно хорошо изученная область микробного метаболизма. Например, для метаболических ферментов информация о том, как мутации влияют на кинетические параметры и как эти параметры различаются между ферментами разных видов, систематически собирается в таких базах данных, как BRENDA.Этот тип информации может быть использован для создания улучшенных предикторов эффекта вариантов специально для метаболических ферментов.
4.2. Моделирование и высокопроизводительный анализ данных
Улучшения в предсказании эффекта варианта в масштабе всего генома также могут быть достигнуты за счет улучшения или расширения подходов к моделированию в масштабе генома. Недавние инновации, такие как GEM-PRO, как обсуждалось в разделе 3.2, удовлетворяют требованиям трехмерных белковых структур для прогнозирования эффектов генетической изменчивости на уровне белка и могут использоваться для систематического анализа влияния генетической изменчивости на метаболизм в масштабе генома. .
Приблизительно 10–30% генов, закодированных в микробном геноме, представлены в метаболических GSM, что ограничивает полезность этих моделей для интерпретации данных о геномных вариантах. Метаболические GSM могут быть расширены несколькими способами, чтобы увеличить охват всего набора генов. Сеть регуляции транскрипции, представленная как взаимодействие между факторами транскрипции и генами-мишенями, может помочь расширить охват прогностических моделей и может быть интегрирована с метаболическими GSM различными способами (Covert et al., 2004; Чандрасекаран и Прайс, 2010). Эти интегрированные модели были успешно использованы для предсказания фенотипов.
Другим недавним расширением GSM являются модели ME. Эти модели учитывают весь механизм, необходимый для экспрессии генов и белков, обеспечивая более широкий охват клеточных функций и более высокое разрешение клеточного состава (O’Brien et al., 2013). МЭ-модели также были расширены, чтобы включить транслокацию белков из цитоплазмы в периплазму (Liu et al., 2014). В настоящее время большинство этих расширений GSM были разработаны только для E. coli , и потребуются значительные усилия для создания этих расширенных моделей для других бактерий, а также эукариотических модельных организмов, таких как S. cerevisiae .
Разработка точных кинетических моделей метаболизма, которые могли бы быть полезны для изучения эффектов мутаций на аллостерическую регуляцию и каталитическую активность, все еще является утомительным процессом. Эти модели обычно ограничиваются небольшими частями метаболизма, фокусируясь на центральном углеродном метаболизме (Chassagnole et al., 2002; Песков и др., 2012; Мачадо и др., 2014). Есть две основные причины этих ограничений: модели становятся огромными по размеру, а кинетическая информация о многих ферментах до сих пор неизвестна. Протоколы (Stanford et al., 2013) и методологии (Chowdhury et al., 2014) разрабатываются, чтобы довести кинетическое моделирование до уровня генома, но полученные модели еще не достигли достаточно зрелой стадии для использования в прогнозировании эффектов вариантов.
На комплексном уровне была предложена стратегия построения моделей целых клеток путем объединения нескольких отдельных моделей различных клеточных процессов, включая клеточный цикл, метаболизм, транскрипцию и транспорт (Karr et al., 2012). Эта стратегия, которая также позволяет комбинировать модели с использованием различных представлений (на основе ограничений, кинетических и стохастических), была использована для построения функционирующей модели целой клетки одного из простейших прокариот, Mycoplasma genitalium . Усилия по созданию более полных моделей микробов в масштабе генома будут продолжаться по мере сбора все большего количества информации и увеличения вычислительной мощности. Эти модели приблизят нас к цели полногеномного предсказания фенотипов на основе данных генотипирования.
4.3. Возможности
Инструменты генной инженерии, такие как MAGE (Wang et al., 2009) или CRISPR/Cas9 (Xu et al., 2014), уже позволяют нам быстро и точно редактировать геномы на уровне разрешения одной пары оснований. в нескольких локусах одновременно. Эти методы позволят нам более полно, чем раньше, картировать эпистатические взаимодействия вариантов внутри одного гена и между несколькими генами. С другой стороны, новые инструменты in silico для прогнозирования влияния вариантов на фенотипы, описанные выше, открывают путь к новому стилю моделирования в масштабе отдельных нуклеотидов.Эти новые инструменты моделирования значительно выиграют от более качественных обучающих наборов данных, которые можно систематически получать с помощью MAGE, CRISPR/Cas9 или других методов редактирования генома для картирования эпистатических взаимодействий. Применение этих новых стратегий обеспечивает способ тонкой настройки активности белков в контексте полных клеточных сетей. Например, мы предвидим, что в будущем у нас будут прогностические модели того, как разработка нескольких ферментов на уровне одной аминокислоты повлияет на производство желаемого метаболита.
Для достижения максимального потенциала моделирования биохимических сетей в масштабе генома и анализа генетических вариантов необходимо установить связь между этими двумя областями. Необходимая информация для соединения обоих миров уже есть: мы знаем гены, белки и реакции. Основные ограничения заключаются в существующих методах и источниках данных. С одной стороны, мы должны преодолеть ограничения инструментов, доступных для прогнозирования эффектов вариантов, позволяя более детально прогнозировать, как вариант может влиять на любую данную функцию или экспрессию белка.Использование предсказаний фолдинга белка, например, уже установлено в метаболическом моделировании (Chang et al., 2013), и должна быть возможность использовать инструменты, которые предсказывают различные эффекты на стабильность белка вместе с моделями масштаба генома. С другой стороны, нам необходимо улучшить методы моделирования биохимических сетей: это развивающаяся область, и в последнее десятилетие были предприняты усилия по стандартизации построения моделей (Thiele and Palsson, 2010) и совершенствованию методов прогнозирования за счет включения высокопроизводительных методов. данные (Machado and Herrgård, 2014).
Наконец, следует признать, что использование исключительно данных о геномных вариантах в качестве основы для предсказания фенотипа для конкретных штаммов всегда будет иметь ограничения. Нам также может понадобиться измерить промежуточные фенотипы, такие как уровни транскриптов, белков или метаболитов для этих штаммов, чтобы сделать прогноз того, как данный генотип влияет на конкретный фенотип (Burga and Lehner, 2013). К счастью, в настоящее время собираются достаточно полные мультиомные наборы данных для микробных штаммов дикого типа, что позволяет усовершенствовать моделирующие и биоинформационные подходы для прогнозирования фенотипа (Ishii et al., 2007; Скелли и др., 2013). Надеемся, что систематизация таких наборов данных и согласованные действия разработчиков моделей, генетиков, микробиологов и биоинформатиков позволят нам прогнозировать измененные и новые метаболические способности микробного штамма на основе данных повторного геномного секвенирования.
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Благодарности
JC, MH и NS признают поддержку Фонда Ново Нордиск через Центр биоустойчивости Фонда Ново Нордиск. Массачусетс признает получение финансирования от гранта на биотехнологические исследования в области синтеза и производства от Фонда Ново Нордиск.
Сноски
Каталожные номера
Аджубей И.А., Шмидт С., Пешкин Л., Раменский В.Е., Герасимова А., Борк П. и др. (2010). Метод и сервер для прогнозирования повреждающих миссенс-мутаций. Нац. Методы 7, 248–249. doi:10.1038/nmeth0410-248
Полнотекстовая перекрестная ссылка | Академия Google
Асадоллахи, М. А., Мори, Дж., Патил, К. Р., Шалк, М., Кларк, А., и Нильсен, Дж. (2009). Повышение производства сесквитерпена в Saccharomyces cerevisiae с помощью метаболической инженерии, управляемой in silico. Метаб. англ. 11, 328–334. doi:10.1016/j.ymben.2009.07.001
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Атсуми, С., Wu, T.-Y., Machado, I.M.P., Huang, W.-C., Chen, P.-Y., Pellegrini, M., et al. (2010). Эволюционный геномный анализ и реконструкция толерантности к изобутанолу у Escherichia coli . Мол. Сист. биол. 6, 449. doi:10.1038/msb.2010.98
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Баренбойм, М., Массо, М., Вайсман, И.И., и Джеймисон, округ Колумбия (2008). Основанное на статистической геометрии предсказание несинонимичных функциональных эффектов SNP с использованием случайного леса и нейро-нечетких классификаторов. Белки 71, 1930–1939. дои: 10.1002/прот.21838
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Брочадо А.Р., Андреев С., Маранас С.Д. и Патил К.Р. (2012). Влияние представления стехиометрии на моделирование отношений генотип-фенотип в метаболических сетях. Вычисление PLoS. биол. 8:e1002758. doi:10.1371/journal.pcbi.1002758
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Брочадо, А.Р., Матос, К., Мёллер, Б.Л., Хансен, Дж., Мортенсен, У.Х., и Патил, К.Р. (2010). Улучшенное производство ванилина в пекарских дрожжах за счет конструкции in silico. Микроб. Сотовый факт. 9, 84. doi:10.1186/1475-2859-9-84
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Бурга, А., и Ленер, Б. (2013). Прогнозирование фенотипической изменчивости по фенотипам генотипов и их комбинации. Курс. мнение Биотехнолог. 24, 803–809.doi:10.1016/j.copbio.2013.03.004
Полнотекстовая перекрестная ссылка | Академия Google
Калабрезе Р., Каприотти Э., Фариселли П., Мартелли П. Л. и Касадио Р. (2009). Функциональные аннотации улучшают прогностическую оценку мутаций белков, связанных с заболеваниями человека. Гул. Мутат. 30, 1237–1244. doi:10.1002/humu.21047
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Каприотти, Э., Калабрезе, Р.и Касадио, Р. (2006). Прогнозирование всплеска генетических заболеваний человека, связанных с точечными белковыми мутациями, с помощью машин опорных векторов и эволюционной информации. Биоинформатика 22, 2729–2734. doi: 10.1093/биоинформатика/btl423
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Caspeta, L., Chen, Y.P., Ghiaci, A.F., Buskov, S., Hallstrom, B.M., Petranovic, D., et al. (2014). Измененный состав стеролов делает дрожжи термоустойчивыми. Наука 346, 75–78. doi:10.1126/наука.1258137
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Чандрасекаран, С., и Прайс, Н.Д. (2010). Вероятностное интегративное моделирование метаболических и регуляторных сетей в масштабе генома у Escherichia coli и Mycobacterium tuberculosis . Проц. Натл. акад. науч. США 107, 17845–17850. doi:10.1073/pnas.1005139107
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Чанг, Р.Л., Эндрюс К., Ким Д., Ли З., Годзик А. и Палссон Б. О (2013). Структурно-системная биология оценки метаболической термотолерантности у Escherichia coli . Наука 340, 1220–1223. doi:10.1126/наука.1234012
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Chassagnole, C., Noisommit-Rizzi, N., Schmid, J.W., Mauch, K., and Reuss, M. (2002). Динамическое моделирование центрального углеродного метаболизма Escherichia coli . Биотехнология. биоинж. 79, 53–73. дои: 10.1002/бит.10288
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Коверт, М. В., Найт, Э. М., Рид, Дж. Л., Херргард, М., и Палссон, Б. Ø (2004). Интеграция высокопроизводительных и вычислительных данных объясняет бактериальные сети. Природа 429, 92–96. дои: 10.1038/природа02456
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Феррер-Коста, К., Gelpi, J.L., Zamakola, L., Parraga, I., de la Cruz, X., and Orozco, M. (2005). PMUT: веб-инструмент для аннотации патологических мутаций белков. Биоинформатика 21, 3176–3178. doi: 10.1093/биоинформатика/bti486
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Голдгар Д., Истон Д., Деффенбо А., Монтейро А., Тавтигян С. и Коуч Ф. (2004). Комплексная оценка вариантов последовательностей ДНК неизвестного клинического значения: приложение к BRCA1 и BRCA2. утра. Дж. Хам. Жене. 75, 535–544. дои: 10.1086/424388
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Гордон, Дж. Л., Бирн, К. П., и Вулф, К. Х. (2009). Дополнения, потери и перестройки на эволюционном пути от реконструированного предка до современного генома Saccharomyces cerevisiae . Генетика PLoS. 5:e1000485. doi:10.1371/journal.pgen.1000485
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Генрих Р.и Шустер, С. (1996). Положение о сотовых системах . Бостон, Массачусетс: Спрингер. дои: 10.1007/978-1-4613-1161-4
Полнотекстовая перекрестная ссылка | Академия Google
Херргард, М., и Панагиоту, Г. (2012). Анализ геномной изменчивости факта микробной клетки в эпоху новых биотехнологий. Вычисл. Структура Биотехнолог. Дж. 3, 1–8. дои: 10.5936/csbj.201210012
Полнотекстовая перекрестная ссылка | Академия Google
Исии, Н., Nakahigashi, K., Baba, T., Robert, M., Soga, T., Kanai, A., et al. (2007). Многочисленные высокопроизводительные анализы отслеживают реакцию E. coli на возмущения. Наука 316, 593–597. doi:10.1126/наука.1132067
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Карчин Р., Диханс М., Келли Л., Томас Д. Дж., Пипер У., Эсвар Н. и др. (2005). LS-SNP: крупномасштабная аннотация кодирования несинонимичных SNP на основе нескольких источников информации. Биоинформатика 21, 2814–2820. doi: 10.1093/биоинформатика/bti442
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Karr, J.R., Sanghvi, J.C., Macklin, D.N., Gutschow, M.V., Jacobs, J.M., Bolival, B., et al. (2012). Вычислительная модель цельной клетки предсказывает фенотип по генотипу. Сотовый 150, 389–401. doi:10.1016/j.cell.2012.05.044
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Келли, Дж.Л., Пейтон, Дж. Т., Фистон-Лавье, А.-С., Титс, Н.М., Йи, М.-К., Джонстон, Дж.С., и соавт. (2014). Компактный геном антарктической мошки, вероятно, является адаптацией к экстремальным условиям. Нац. коммун. 5, 4611. doi:10.1038/ncomms5611
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Кирхер, М., Виттен, Д.М., Джайн, П., О’Роак, Б.Дж., Купер, Г.М., и Шендуре, Дж. (2014). Общая основа для оценки относительной патогенности генетических вариантов человека. Нац. Жене. 46, 310–315. дои: 10.1038/ng.2892
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Косури, С., Гудман, Д. Б., Камбрей, Г., Муталик, В. К., Гао, Ю., Аркин, А. П., и соавт. (2013). Компоновка регуляторных последовательностей, контролирующих транскрипцию и трансляцию в Escherichia coli . Проц. Натл. акад. науч. США 110, 14024–14029. doi:10.1073/pnas.1301301110
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ли, Дж., Юн, Х., Файст, А.М., Палссон, Б.О, и Ли, С.Ю. (2008). Реконструкция в масштабе генома и анализ in silico метаболической сети Clostridium acetobutylicum ATCC 824. Заяв. микробиол. Биотехнолог. 80, 849–862. дои: 10.1007/s00253-008-1654-4
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ли, Б., Кришнан, В.Г., Морт, М.Е., Синь, Ф., Камати, К.К., Купер, Д.Н., и соавт. (2009). Автоматический вывод о молекулярных механизмах заболевания по аминокислотным заменам. Биоинформатика 25, 2744–2750. doi: 10.1093/биоинформатика/btp528
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ли, Р., Фань, В., Тиан, Г., Чжу, Х., Хе, Л., Цай, Дж., и др. (2010). Последовательность и сборка de novo генома гигантской панды. Природа 463, 311–317. дои: 10.1038/nature08696
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Лю, Дж.К., О’Брайен, Э. Дж., Лерман, Дж. А., Зенглер, К., Палссон, Б. О, и Файст, А. М. (2014). Реконструкция и моделирование транслокации и компартментализации белка в Escherichia coli в масштабе генома. BMC Сист. биол. 8:110. doi: 10.1186/s12918-014-0110-6
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Меламед, Д., Янг, Д.Л., Гэмбл, К.Е., Миллер, Ч.Р., и Филдс, С. (2013). Глубокое мутационное сканирование домена RRM поли(А)-связывающего белка Saccharomyces cerevisiae . РНК 19, 1537–1551. doi:10.1261/РНК.040709.113
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Накамура Ю., Мори К., Сайто К., Осима К., Мекути М., Сугая Т. и др. (2013). Эволюционные изменения нескольких генов зрительного пигмента в полном геноме тихоокеанского голубого тунца. Проц. Натл. акад. науч. США 110, 11061–11066. doi:10.1073/pnas.1302051110
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Нам, Х., Campodonico, M., Bordbar, A., Hyduke, D.R., Kim, S., Zielinski, D.C., et al. (2014). Системный подход к прогнозированию онкометаболитов с помощью контекстно-специфических метаболических сетей в масштабе генома. Вычисление PLoS. биол. 10:e1003837. doi:10.1371/journal.pcbi.1003837
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
О’Брайен, Э. Дж., Лерман, Дж. А., Чанг, Р. Л., Хайдук, Д. Р., и Палссон, Б. О (2013). Геномные модели метаболизма и экспрессии генов расширяют и уточняют предсказание фенотипа роста. Мол. Сист. биол. 9, 693–693. doi: 10.1038/msb.2013.52
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Оксер С., Пахиккала Т., Айрола А., Салакоски Т., Рипатти С. и Айттокаллио Т. (2014). Регуляризованное машинное обучение в генетическом прогнозировании сложных признаков. Генетика PLoS. 10:e1004754. doi:10.1371/journal.pgen.1004754
Полнотекстовая перекрестная ссылка | Академия Google
Орт, Дж.Д., Тиле И. и Палссон Б. О (2010). Что такое анализ баланса потоков? Нац. Биотехнолог. 28, 245–248. doi:10.1038/nbt.1614
Полнотекстовая перекрестная ссылка | Академия Google
Парк, Дж. Х., Ли, К. Х., Ким, Т. Ю., и Ли, С. Ю. (2007). Метаболическая инженерия Escherichia coli для производства L-валина на основе анализа транскриптома и моделирования нокаута генов in silico. Проц. Натл. акад. науч. США 104, 7797–7802.doi:10.1073/pnas.0702609104
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Puchałka, J., Oberhardt, M.A., Godinho, M., Bielecka, A., Regenhardt, D., Timmis, K.N., et al. (2008). Реконструкция в масштабе генома и анализ метаболической сети Pseudomonas putida KT2440 облегчают применение в биотехнологии. Вычисление PLoS. биол. 4:e1000210. doi:10.1371/journal.pcbi.1000210
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Шварц, Дж.М., Купер Д.Н., Шуэльке М. и Зеелов Д. (2014). MutationTaster2: предсказание мутаций для возраста глубокого секвенирования. Нац. Методы 11, 361–362. doi:10.1038/nmeth.2890
Полнотекстовая перекрестная ссылка | Академия Google
Shihab, H.A., Gough, J., Cooper, D.N., Stenson, P.D., Barker, G.L.A., Edwards, K.J., et al. (2012). Прогнозирование функциональных молекулярных и фенотипических последствий аминокислотных замен с использованием скрытых марковских моделей. Гул.Мутат. 34, 57–65. doi:10.1002/humu.22225
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Скелли, Д. А., Меррихью, Г. Е., Риффл, М., Коннелли, С. Ф., Керр, Э. О., Йоханссон, М., и соавт. (2013). Интегративная феномика дает представление о структуре фенотипического разнообразия почкующихся дрожжей. Рез. генома. 23, 1496–1504. дои: 10.1101/гр.155762.113
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Сниткин Е.С., Дадли А.М., Янсе Д.М., Вонг К., Черч Г.М. и Сегре Д. (2008). Управляемый моделью анализ экспериментально определенных фенотипов роста для 465 мутантов с делецией гена дрожжей в 16 различных условиях. Геном Биол. 9, Р140. doi: 10.1186/gb-2008-9-9-r140
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Стэнфорд, штат Нью-Джерси, Любиц, Т., Смоллбоун, К., Клипп, Э., Мендес, П., и Либермейстер, В. (2013). Систематическое построение кинетических моделей из метаболических сетей геномного масштаба. PLoS One 8:e79195. doi:10.1371/journal.pone.0079195
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Шаппанос Б., Ковач К., Шамеч Б., Хонти Ф., Костанцо М., Барышникова А. и др. (2011). Комплексный подход к характеристике сетей генетических взаимодействий в метаболизме дрожжей. Нац. Жене. 43, 656–662. дои: 10.1038/ng.846
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Терп, Б.Н., Купер Д.Н., Кристенсен И.Т., Йогенсен Ф.С., Бросс П., Грегерсен Н. и соавт. (2002). Оценка относительной важности биофизических свойств аминокислотных замен, связанных с генетическими заболеваниями человека. Гул. Мутат. 20, 98–109. doi:10.1002/humu.10095
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Thiele, I., Swainston, N., Fleming, R.M., Hoppe, A., Sahoo, S., Aurich, M.K., et al. (2013).Глобальная реконструкция метаболизма человека, управляемая сообществом. Нац. Биотехнолог. 31, 419–425. doi:10.1038/nbt.2488
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Томас, П. Д., и Кеджаривал, А. (2004). Кодирование однонуклеотидных полиморфизмов, связанных со сложным заболеванием по сравнению с болезнью Менделя: эволюционные доказательства различий в молекулярных эффектах. Проц. Натл. акад. науч. США 101, 15398–15403. дои: 10.1073/пнас.0404380101
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Тянь Дж., Ву Н., Го X., Го Дж., Чжан Дж. и Фань Ю. (2007). Прогнозирование фенотипических эффектов несинонимичных однонуклеотидных полиморфизмов на основе машин опорных векторов. Биоинформатика BMC 8:450. дои: 10.1186/1471-2105-8-450
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Варма, А.и Палссон, Б. О (1993). Метаболические возможности Escherichia coli II. оптимальные схемы роста. Ж. Теор. биол. 165, 503–522. дои: 10.1006/jtbi.1993.1203
Полнотекстовая перекрестная ссылка | Академия Google
Wang, H.H., Isaacs, FJ, Carr, P.A., Sun, Z.Z., Xu, G., Forest, C.R., et al. (2009). Программирование клеток с помощью мультиплексной инженерии генома и ускоренной эволюции. Природа 460, 894–898. дои: 10.1038/природа08187
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ван, Л., Han, X., Zhang, Y., Li, D., Wei, X., Ding, X., et al. (2014). Глубокое повторное секвенирование выявило аллельные вариации Sesamum indicum. BMC Растение Биол. 14:225. doi:10.1186/s12870-014-0225-3
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ван, М., Чжао, X., Такемото, К., Сюй, Х., Ли, Ю., Акуцу, Т., и др. (2012). FunSAV: прогнозирование функционального эффекта вариантов одной аминокислоты с использованием двухэтапной модели случайного леса. PLoS One 7:e43847.doi:10.1371/journal.pone.0043847
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Сюй Т., Ли Ю., Ностранд Дж. Д. В., Хе З. и Чжоу Дж. (2014). Инструменты на основе Cas9 для целенаправленного редактирования генома и контроля транскрипции. Заяв. Окружающая среда. микробиол. 80, 1544–1552. doi: 10.1128/AEM.03786-13
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ямамото К., Тамаки Х., Кадильо-Кирос Х., Имачи Х., Кирпидес Н., Войке Т. и др. (2014). Полная последовательность генома Methanoregula formicica SMSPT, мезофильного гидрогенотрофного метаногена, выделенного из метаногенного анаэробного реактора с защитным слоем ила с восходящим потоком. Объявление генома. 2, е870–е814. doi:10.1128/genomeA.00870-14
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ян Х., Крумхольц Э. В., Брутинель Э. Д., Палани Н.П., Садовски М.Дж., Одлызко А.М. и соавт. (2014). Валидация метаболической сети в масштабе генома Shewanella oneidensis с использованием анализа частоты вставки транспозона. Вычисление PLoS. биол. 10:e1003848. doi:10.1371/journal.pcbi.1003848
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Ye, Z.-Q., Zhao, S.-Q., Gao, G., Liu, X.-Q., Langlois, R.E., Lu, H., et al. (2007). Поиск новых структурных и последовательных атрибутов для прогнозирования возможной ассоциации полиморфизма одной аминокислоты (SAP) с заболеванием. Биоинформатика 23, 1444–1450. doi: 10.1093/биоинформатика/btm119
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Чжао, Н., Хан, Дж. Г., Шью, К.-Р., и Коркин, Д. (2014). Определение влияния несинонимичных SNP на белок-белковые взаимодействия с использованием контролируемого и полуконтролируемого обучения. Вычисление PLoS. биол. 10:e1003592. doi:10.1371/journal.pcbi.1003592
Опубликовано Резюме | Опубликован полный текст | Полный текст перекрестной ссылки | Академия Google
Генетическая изменчивость в популяции
Генетическая изменчивость в популяции описывает наличие в этой популяции различных аллелей или альтернативных форм для данного гена.Наличие генетической изменчивости означает, что особи популяции различаются по аллелям. они обладают, что означает, что люди различаются по генотипу. Генетические локусы, для которых существует несколько аллелей, описываются как полиморфные. Люди, например, полиморфны по таким признакам, как цвет глаз и группа крови.
Генетическая изменчивость является одним из аспектов более общей концепции фенотипической изменчивости. Фенотипическая изменчивость описывает различия в характеристиках особей популяции.Фенотипическая изменчивость представляет интерес для биологов, потому что именно на нее действует естественный отбор: разные фенотипы могут иметь разную приспособленность, и в результате отбора более приспособленные фенотипы оставляют больше потомков.
Фенотипическая изменчивость возникает из двух источников: генетической изменчивости и изменчивости окружающей среды. Однако только те различия, которые возникают в результате генетической изменчивости, могут быть переданы будущим поколениям. Более того, только часть генетического компонента изменчивости, аддитивная генетическая изменчивость, действительно наследуема.Аддитивная генетическая изменчивость, деленная на общую фенотипическую изменчивость, дает наследуемость, которая описывает, насколько потомство похоже на своих родителей.
Количество генетических вариаций
В 1960-х годах велись серьезные споры о том, сколько генетических вариаций действительно существует в популяциях. Принято считать, что полиморфные локусы встречаются довольно редко. Затем разработка техники гель-электрофореза позволила биологам изучить закономерности изменения белков в разных популяциях и количественно оценить генетическую изменчивость.
Биологи обнаружили удивительно большое количество генетических вариаций. Например, у большинства видов позвоночных примерно 30% генов оказались полиморфными. Исследования, проведенные в 1970-х годах на людях, показали, что генетическая изменчивость встречается примерно на том же уровне, что и у других видов животных. Исследования на людях также показали, как известно, что так называемые человеческие расы не являются реальными биологическими группами. Было обнаружено, что генетическая изменчивость внутри рас значительно больше, чем между ними.
С тех пор отсутствие генетических вариаций считается аномальным. Отсутствие генетической изменчивости в популяциях обычно предполагает, что в недавней истории группы было узкое место в популяции, когда размер популяции стал очень маленьким. Результатом узкого места в популяции является то, что все члены нынешней популяции происходят от небольшого числа особей и, следовательно, имеют лишь ограниченную генетическую изменчивость. Ожидается, что генетическая изменчивость со временем будет накапливаться в этих популяциях по мере появления новых мутаций.
Как поддерживается генетическая изменчивость
Открытие большого количества генетической изменчивости почти во всех популяциях привело к постановке другого вопроса: как поддерживается генетическая изменчивость? В конце концов, во многих случаях естественный отбор устраняет генетическую изменчивость, устраняя менее приспособленные генотипы.
Многие факторы увеличивают или поддерживают количество генетических вариаций в популяции. Одним из них является мутация, которая фактически является основным источником всех вариаций.Однако мутации происходят не очень часто, только со скоростью примерно одна мутация на 100 000–1 000 000 генетических локусов в поколении. Эта скорость слишком мала, чтобы объяснить большую часть полиморфизмы, встречающиеся в природных популяциях. Тем не менее, мутация, вероятно, объясняет некоторые очень редкие фенотипы, наблюдаемые время от времени, такие как альбинизм у людей и других млекопитающих.
Вторым фактором, способствующим генетической изменчивости природных популяций, является избирательная нейтральность. Селективная нейтральность описывает ситуации, в которых альтернативные аллели гена мало отличаются по приспособленности.Поскольку небольшие различия в приспособленности приводят лишь к слабому естественному отбору, отбор может быть подавлен случайной силой генетического дрейфа. Аллели, частоты которых определяются генетическим дрейфом, а не естественным отбором, называются избирательно нейтральными. При нейтральности частоты аллелей меняются со временем, случайным образом увеличиваясь или уменьшаясь. В течение длительных периодов времени случайные колебания относительной частоты различных аллелей могут привести к тому, что некоторые из них исчезнут из популяции.Однако генетические полиморфизмы долгоживущие, и новые нейтральные аллели могут постоянно возникать в результате мутаций.
Наконец, некоторые формы естественного отбора скорее поддерживают генетическую изменчивость, чем устраняют ее. К ним относятся уравновешивающий отбор, частотно-зависимый отбор и изменение закономерностей естественного отбора во времени и пространстве.
Балансирующий отбор имеет место, когда в локусе имеется преимущество гетерозигот, ситуация, при которой гетерозиготный генотип (один включает два разных аллеля) имеет большую приспособленность, чем любой из двух гомозиготных генотипов (один включает два одинаковых аллеля).При преимуществе гетерозигот оба задействованных аллеля будут сохраняться в популяции.
Классический пример преимущества гетерозигот касается аллеля серповидноклеточной анемии. Лица, гомозиготные по аллелю серповидноклеточной анемии, имеют серповидноклеточную анемию, которая вызывает изменение эритроцитов. серповидной формы, когда они выделяют кислород. Эти серповидные клетки попадают в узкие кровеносные сосуды, блокируя кровоток. До разработки современных методов лечения заболевание ассоциировалось с очень низкой приспособленностью, поскольку люди обычно умирали до репродуктивного возраста.
Гетерозиготы, однако, имеют нормальные клетки крови в форме пончика и не страдают серповидноклеточной анемией. Кроме того, они пользуются преимуществом серповидноклеточного аллеля, который обеспечивает защиту от малярии. Следовательно, гетерозиготные особи обладают большей приспособленностью, чем особи, имеющие две копии нормального аллеля. Считается, что преимущество гетерозигот в этой системе сыграло решающую роль в сохранении в человеческом населении такого опасного заболевания, как серповидноклеточная анемия.Доказательства этого исходят из изучения распространения аллеля серповидноклеточной анемии, который встречается только в местах, где малярия представляет опасность.
Другой формой естественного отбора, которая поддерживает генетическую изменчивость в популяциях, является частотно-зависимый отбор. При частотно-зависимом отборе приспособленность генотипа зависит от его относительной частоты в популяции, при этом менее распространенные генотипы более подходят, чем генотипы, встречающиеся с высокой частотой.
Считается, что частотно-зависимый отбор довольно распространен в природных популяциях.Например, в ситуациях конкуренции за ресурсы люди с редкими предпочтениями могут обладать большей приспособленностью, чем те, у кого общие предпочтения. Частотно-зависимый отбор также может играть роль в хищничестве: если хищники формируют образ поиска более распространенных типов добычи, сосредоточив внимание на их поимке, менее распространенные фенотипы могут лучше выжить.
Наконец, изменение моделей отбора во времени или пространстве может помочь сохранить генетическую изменчивость в популяции.Если модели отбора со временем меняются, разные аллели или генотипы могут обладать большей приспособленностью в разное время. Общий эффект может заключаться в том, что оба аллеля сохраняются в популяции. Изменяющееся давление отбора с течением времени встречается у видов кузнечиков, характеризующихся двумя цветовыми морфами: коричневой морфой и зеленой морфой. В начале года, когда среда обитания более коричневая, лучше замаскированные коричневые кузнечики пользуются большей защитой от хищников. Однако в конце сезона окружающая среда становится зеленее, а зеленые кузнечики более приспособлены.
Другая возможность заключается в том, что модели отбора варьируются от одного места к другому в результате различий в среде обитания и окружающей среде. Преобладание разных генотипов в разных местах обитания в сочетании с потоком генов между местами обитания может привести к сохранению множественных аллелей в популяции.
Одним из примеров является аллель устойчивости к токсичности меди у видов трав. Аллели толерантности к меди распространены в районах, прилегающих к медным рудникам, где почва загрязнена.Однако они не ожидаются в незагрязненных районах, где они менее приспособлены, чем нормальные аллели. Однако, поскольку виды трав опыляются ветром, гаметы могут перемещаться на значительные расстояния, а аллели, устойчивые к меди, часто обнаруживаются в районах, где они находятся в невыгодном положении.
см. также Гены; генетика; Перцовая моль; Селекционное скрещивание.
Дженнифер Йе
Библиография
Кертис, Хелена. Биология. Нью-Йорк: издательство Worth, 1989.
Футуйма, Дуглас Дж. Эволюционная биология. Сандерленд, Массачусетс: Sinauer Associates, 1998.
Гулд, Джеймс Л. и Уильям Т. Китон, с Кэрол Грант Гулд. Биологические науки, 6-е изд. Нью-Йорк: WW Norton & Company, 1996.
Паттерсон, Колин. Эволюция, 2-е изд. Итака, Нью-Йорк: Комсток, 1999.
Ридли, Марк. Эволюция. Boston: Blackwell Scientific, 1993.
Одним из хорошо изученных примеров генетической изменчивости популяций является Biston betularia, перечная моль.
У перечной моли есть три цветовые морфы: светлая морфа, темная или меланистическая морфа и промежуточная морфа. До промышленной революции наиболее распространенной формой была светлая морфа, хотя иногда встречались и меланистические бабочки. Однако к концу девятнадцатого века меланистическая морфа стала гораздо более распространенной, а в некоторых областях практически вытеснила светлую морфу.
Биологи связали этот сдвиг с промышленным загрязнением городских территорий.Без замаскированных мест отдыха легкие бабочки становились легкой мишенью для хищников-птиц. Этим объяснялось как преобладание меланистических бабочек в загрязненных городских условиях, так и светлых бабочек в сравнительно нетронутых сельских местообитаниях.
Загадочный аспект истории с перцовой бабочкой заключается в том, что генетическая изменчивость не была полностью устранена в популяциях. В городах, например, бабочки-меланисты составляют лишь от 90 до 100% общей популяции, несмотря на очень сильный отбор.По-видимому, действуют и другие силы, помимо давления хищников. На короткое время было высказано предположение, что объяснением может быть преимущество гетерозигот, но в конечном итоге эта теория была отвергнута. В настоящее время считается, что поток генов между деревней и городскими районами и частотно-зависимый отбор являются жизнеспособными альтернативами. Однако предстоит еще много работы над этой исторической системой.
АКВА | Биология | Содержание темы
Биологическое разнообразие – биоразнообразие – выражается в огромном количестве видов организмов, в изменчивости индивидуальных признаков в пределах одного вида и в разнообразии типов клеток в пределах одного многоклеточного организма.
Различия между видами отражают генетические различия. Различия между особями внутри вида могут быть результатом генетических факторов, факторов окружающей среды или их комбинации.
Ген представляет собой участок ДНК, расположенный в определенном месте молекулы ДНК, называемом его местонахождение. Базовая последовательность каждого гена несет закодированную генетическую информацию, которая определяет последовательность аминокислот при синтезе белка.Используемый генетический код одинаков во всех организмов, что косвенно свидетельствует об эволюции.
Генетическое разнообразие внутри вида может быть вызвано генной мутацией, хромосомной мутацией или случайными факторами, связанными с мейозом и оплодотворением. На это генетическое разнообразие действует естественный отбор, в результате чего виды лучше адаптируются к окружающей среде.
Изменчивость внутри вида можно измерить, используя различия в базовой последовательности ДНК или в аминокислотной последовательности белков.
Биоразнообразие в сообществе можно измерить с помощью видового богатства и индекса разнообразия.
ДНК, гены и хромосомы
| Содержание |
Возможности для развития навыков |
|---|---|
|
В прокариотических клетках молекулы ДНК короткие, кольцевые. и не связаны с белками. В ядре эукариотических клеток молекулы ДНК очень длинные, линейные и связанные с белками, называемыми гистонами. Вместе молекула ДНК и связанные с ней белки образуют хромосому. Митохондрии и хлоропласты эукариотических клеток также содержат ДНК, которая, как и ДНК прокариот, короткая, кольцевая и не связаны с белком. Ген представляет собой базовую последовательность ДНК, которая кодирует:
Ген занимает фиксированное положение, называемое локусом, на конкретной молекулы ДНК. Последовательность из трех оснований ДНК, называемая триплетом, кодирует конкретной аминокислоты. Генетический код универсален, непересекающиеся и вырожденные. У эукариот большая часть ядерной ДНК не кодирует полипептиды. Существуют, например, некодирующие множественные повторы последовательности оснований между генами.Даже внутри гена лишь некоторые последовательности, называемые экзонами, кодируют аминокислотные последовательности. В рамках ген, эти экзоны разделены одной или несколькими некодирующими последовательностями, называются интронами. |
Синтез ДНК и белка
| Содержание |
Возможности для развития навыков |
|---|---|
|
Представление о геноме как о полном наборе генов в клетки и протеома как полного спектра белков, которые клетка умеет производить. Структура молекул информационной РНК (мРНК) и транспортная РНК (тРНК). Транскрипция как получение мРНК из ДНК. Роль РНК полимеразы в соединении нуклеотидов мРНК.
Трансляция как получение полипептидов из последовательность кодонов, переносимых мРНК. Роль рибосом, тРНК и АТФ. Учащиеся должны уметь:
Учащиеся , а не должны будут вспомнить в письменной форме документирует конкретные кодоны и аминокислоты, которые они кодируют. |
Генетическое разнообразие может возникнуть в результате мутации или в ходе мейоза
| Содержание |
Возможности для развития навыков |
|---|---|
|
Генные мутации связаны с изменением базовой последовательности хромосом.Они могут возникать спонтанно во время репликации ДНК и включать в себя удаление и замена оснований. В связи с дегенеративным характером генетическом коде, не все замены оснований вызывают изменение последовательность кодируемых аминокислот. Мутагены могут увеличивать скорость генной мутации. Мутации числа хромосом могут возникать спонтанно в результате нерасхождения хромосом во время мейоза. Мейоз производит дочерние клетки, которые генетически отличаются друг от друга. Процесс мейоза только достаточно подробно, чтобы показать как:
Учащиеся должны уметь:
|
В д
Студенты могли исследовать мейоз на подготовленных предметных стеклах.
подходящей растительной или животной ткани.
МС 0,5
Учащиеся могут:
|
Генетическое разнообразие и адаптация
| Содержание |
Возможности для развития навыков |
|---|---|
|
Генетическое разнообразие как количество различных аллелей генов в популяции. Генетическое разнообразие является фактором, обеспечивающим естественную происходить отбор. Принципы естественного отбора в эволюции населения.
Направленная селекция на примере антибиотиков устойчивость у бактерий и стабилизирующий отбор на примере веса человека при рождении. Естественный отбор приводит к появлению видов, которые лучше приспособлены к своим окружающая обстановка. Эти приспособления могут быть анатомическими, физиологическими или поведенческий. Учащиеся должны уметь:
|
МС 2,5 Студенты могли использовать логарифмическую шкалу при работе с данные о большом количестве бактерий в культуре. |
| Требуемый практический 6: Использование асептических методов исследовать действие антимикробных веществ на микроорганизмы рост. | АТ и |
Виды и таксономия
| Содержание |
Возможности для развития навыков |
|---|---|
| Два организма принадлежат к одному и тому же виду, если они способны производить фертильные потомство. Ухаживание как необходимый предшественник успешное спаривание.Роль ухаживания в распознавании видов. Система филогенетической классификации пытается объединить виды в группы на основе их эволюционного происхождения и взаимоотношений. Он использует иерархию, в которой меньшие группы помещаются в более крупные группы без перекрытия между группами. Каждая группа называется таксоном (множественное число таксонов). Одна иерархия включает таксоны: домен, царство, тип, класс, порядок, семейство, род и вид. Каждый вид повсеместно идентифицируется биномом, состоящим из названия его род и вид, например, Homo sapiens . Вспомнить различные таксономические системы, такие как три системы домена или пяти королевств, потребуется , а не . Учащиеся должны уметь оценить достижения в области иммунологии и секвенирование генома помогает прояснить эволюционные отношения между организмами. |
| Содержание |
Возможности для развития навыков |
|---|---|
| Биоразнообразие может относиться к целому ряду мест обитания, от небольшой местной среды обитания до Земли. Видовое богатство – это мера количества различных видов в сообществе. Индекс разнообразия описывает соотношение между числом видов в сообществе и числом особей каждого вида. Расчет индекса разнообразия ( ) из формулы где = общее количество организмов всех видов и = общее количество организмов каждого вида. Методы ведения сельского хозяйства сокращают биоразнообразие. Баланс между сохранением и сельским хозяйством. |
МС 2.3 Студентам могут быть предоставлены данные для расчета индекс разнообразия и интерпретировать значимость рассчитанного значение индекса. |
Изучение разнообразия
| Содержание |
Возможности для развития навыков |
|---|---|
Генетическое разнообразие внутри видов или между видами можно получить путем сравнения:
Количественные исследования изменчивости в виды включают:
Учащимся потребуется , а не для расчета стандартные отклонения в письменных работах. |
АТ к Студенты могли:
МС 1.2 Учащиеся могут использовать стандартные научные калькуляторы для расчета средних значений данных, которые они собрали или получили. МС 1.10 Учащиеся могли рассчитать и интерпретировать значения стандартного отклонения их средних значений. |
Генетическая изменчивость и естественный отбор: естественный отбор
Естественный отбор
Как обсуждалось в разделе «Структура и функции специализированных клеток», половое размножение и мутация ДНК являются двумя основными процессами, увеличивающими генетическую изменчивость.Хотя мутация является единственным источником новых аллелей, возможность новой комбинации аллелей увеличивается с каждым половым размножением из трех первичных процессов:
- Случайное соединение сперматозоида и яйцеклетки
- Кроссинговер во время мейоза 1
- Независимый набор гомологичных хромосомы
Хотя большинство генетических рекомбинаций с участием рецессивного гена биологически нейтральны, потенциал экспрессии этого гена в будущем все еще сохраняется в этом организме и, следовательно, в этой размножающейся популяции.Увеличение генетической изменчивости и влияние окружающей среды на эту изменчивость составляют основу естественного отбора . Естественный отбор — это теория, утверждающая, что те особи, которые лучше всего приспособлены к жизни в данной местности, выживают и размножаются. Внутри данной популяции существует нормальная степень генетической изменчивости, которая может или не может сделать индивидуума более приспособленным к окружающей среде или, что более важно, к изменениям в окружающей среде. Вид с большой генетической изменчивостью имеет больше шансов выжить как вид в изменяющейся среде, чем вид с ограниченной изменчивостью.
Дарвин и естественный отбор
Теории Чарльза Дарвина до сих пор составляют основу нашего понимания естественного отбора. До сих пор широко распространено мнение, что естественный отбор основан на пяти факторах:
Bionote
Дарвин много писал о галапагосских вьюрках, которых он изучал во время исследовательского рейса на HMS Beagle . Он внимательно наблюдал и отмечал, что различия в строении клюва у вьюрков создали определенные преимущества или различия в их поиске пищи.Например, одни клювы были предназначены для раздавливания семенных оболочек, а другие — для ловли насекомых. Он наиболее тесно связан с изучением вьюрков.
- Внутри вида индивидуальные вариации существуют естественным образом. Некоторые организмы быстрее, красочнее, крупнее или умнее других представителей своего вида. Индивидуальные вариации иногда бывают полезными, нейтральными или вредными.
- Потомство рождается больше, чем может выжить, и их успех в жизненной борьбе за пищу, кров и пару определяет их способность успешно воспроизводить и передавать свой генетический набор.
- Репродуктивные способности особей неодинаковы или благоприятны в зависимости от давления окружающей среды.
- Условия окружающей среды определяют репродуктивный успех определенных особей, потому что они обладают чертой, которая дает им преимущество в этой среде.
- Особи, способные обеспечить себя необходимой пищей, убежищем и избежать нападения хищников, размножаются более успешно, чем другие. На протяжении поколений характеристики популяции меняются по мере того, как особи, более успешные в размножении, заселяют вид.Менее успешные типы не передают столько признаков, потому что у них меньше потомков. Со временем неспособность воспроизводиться со скоростью, равной или превышающей уровень смертности, приводит к вымиранию. Процесс, при котором частота определенных признаков внутри вида изменяется из-за неравномерной скорости размножения, вызванной естественным отбором, называется эволюцией .
Биотермс
Хищничество — отношения между видами, при которых один вид, хищник, поедает другой вид, добычу.
В модели Дарвина естественный отбор не обязательно запрещал индивидууму размножаться, а скорее отдавал предпочтение наиболее приспособленным индивидуумам с большими шансами на размножение, возможностью большего числа рождений и ускоренным развитием и выживанием потомства. Таким образом, естественный отбор позволяет общему количеству определенных членов популяции сокращаться или вымирать из-за давления окружающей среды, в то время как другие соответственно увеличиваются.
Классическое исследование естественного отбора было описано Х.B. D. Kettlewell в 1952 г. В весьма интересном эксперименте он пришел к выводу, что влияние окружающей среды, хищничество птиц, влияет на общее число размножающихся бабочек в зависимости от их окраски. Кеттлуэлл в то время работал в Оксфордском университете и обнаружил обрывок информации 1840-х годов, 100 лет назад, в которой отмечалось первое появление темной морфы перечной бабочки. До этого времени наблюдались только белые или перцовые бабочки. Он связал даты с началом тяжелой промышленности в этом районе.Он также знал, что заводы в то время и в его время ежедневно производили объемные клубы черного дыма, сильно отягощенного сажей. Он также знал, что перечная моль распространена по всей Англии; они вели ночной образ жизни и днем прятались на стволах деревьев; на них охотились многие виды птиц. Он предположил, что птицы охотятся на мотыльков, которые менее замаскированы и поэтому их легче заметить. Поступая таким образом, они предпочитали один тип морфа другому, что создавало неравномерную скорость размножения, что способствовало увеличению количества одного типа мотылька по сравнению с другими.
Bionote
Альфред Уоллес также предложил теорию естественного отбора одновременно с Дарвином; однако имя Дарвина связано с этой идеей, вероятно, из-за большей осведомленности, обеспеченной его книгой «Происхождение видов », опубликованной в 1859 году. темные мотыльки в обычных лесах. Он помечал каждого мотылька мазком краски, а затем устанавливал ловушки для их повторной поимки.В закопченном темном лесу он отловил в основном темных мотыльков; в более светлом лесу в основном светлые мотыльки. Он пришел к выводу, что неравномерные показатели повторной поимки были основаны на неравномерных показателях хищничества. Чтобы подтвердить свои подозрения, он наблюдал и фотографировал птиц, охотящихся на мотыльков, цвет тела которых контрастировал с окружающей средой, при этом явно не замечая более замаскированного мотылька. Те бабочки, которые лучше всего приспособились к окружающей среде, выживали и размножались; другие этого не сделали. Эксперимент Кеттлуэлла является примером того, как давление окружающей среды может определять характеристики видов.
В этот исторический период теории Дарвина встретили сильное противодействие со стороны других ученых и религиозных лидеров из-за их новизны и связанного с ними противоречивого характера.
