
Готовимся к ЕГЭ. Часть I
Верны ли следующие суждения
№1
А. Делением размножаются только клетки, но не органоиды.
Б. Клетки размножаются делением. а вирусы нет.
1. Верно только А
2. Верно только Б
3. Верны обе формулировки
4. Обе формулировки не верны
№2
А. При дигибидном скрещивании у гибридов каждая пара признаков наследуется независимо от других и дает с ними разные сочетания.
Б. Это закон чистоты гамет: в каждую гамету попадает только одна аллель из пары аллелей данного гена родительской особи.
Б. Пары альтернативных признаков не смешиваются и при образовании гамет по одному переходят в них в чистом виде.
№3
А. Результатами эволюции можно считать разнообразие организмов и их приспособленность к условиям окружающей среды.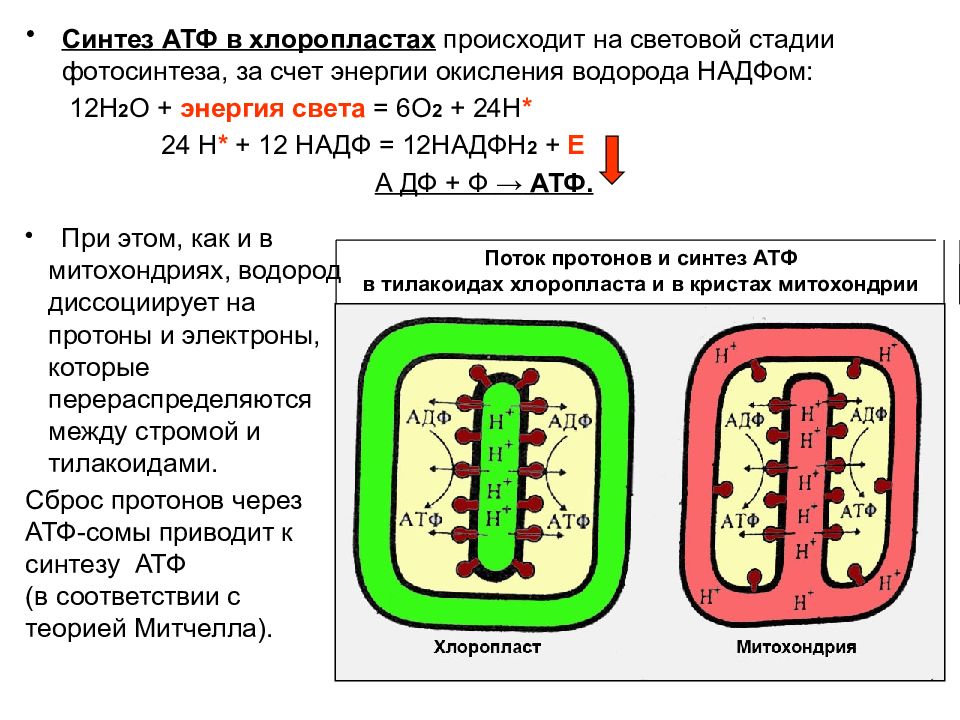
Б. Результатами эволюции считаются изоляция, борьба за существование и возникновение мутаций.
№4
А. Наследственная изменчивость служит материалом для естественного отбора.
Б. Наследственная изменчивость способствует сохранению в популяции наиболее приспособленных особей
№5
А. Многие белки выполняют каталитическую функцию.
Б. Некоторые гормоны имеют белковую природу.
№6
Упрощение в строении животных, связанные с паразитическим образом жизни, относят к биологическому регрессу.
Б. Возникновение класса Насекомые, сопровождающееся повышением нового уровня их организации, — пример ароморфоза.
№7
А. Генетический код триплетен – один триплет всегда кодирует только одну аминокислоту.
Б. Генетический код однозначен – в молекуле нуклеиновой кислоты одна аминокислота кодируется сочетанием трех последовательно расположенных нуклидов.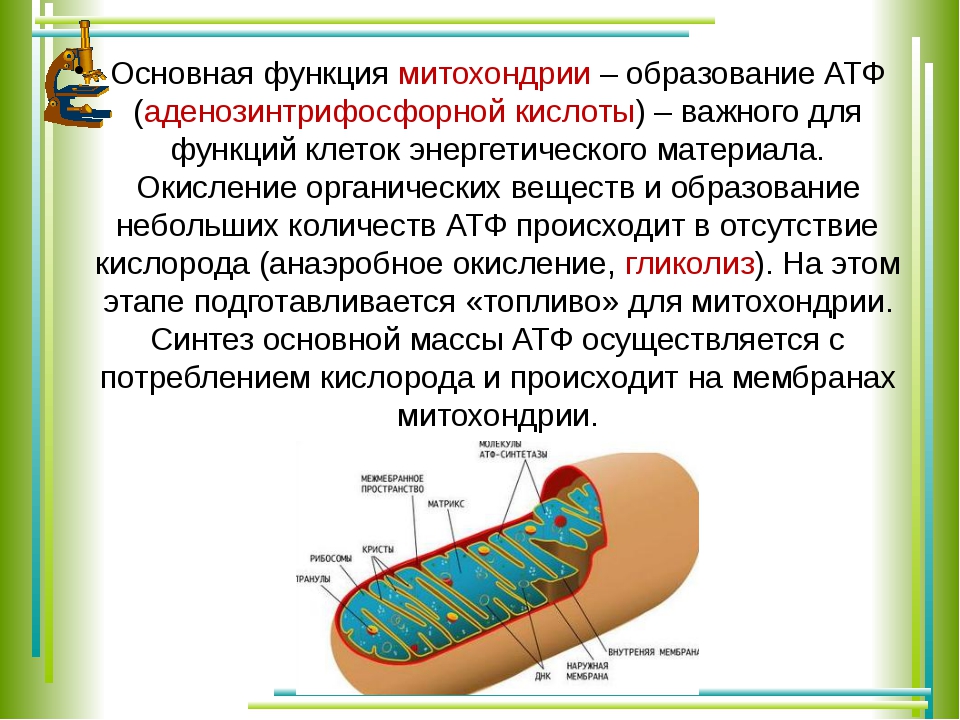
№8
А. В результате митоза из одной диплоидной клетки образуется 4 гоплоидные.
Б. Редукция числа хромосом происходит в анафазе I.
№9
А. Мутационная изменчивость носит случайный характер.
Б. Признак человека «масса тела» обладает узкой нормой реакции.
№10
А. Митоз обеспечивает рост организма, регенерацию, лежит в основе бесполого размножения.
№11
Верны ли следующие суждения о фотосинтезе?
А. В световой фазе фотосинтеза происходит синтез Атор, образуются атомы водорода и молекулярный кислород.
Б. Световая фаза фотосинтеза происходит в строме хлоропласта.
№12
Верны ли следующие суждения о химическом составе клетки?
А.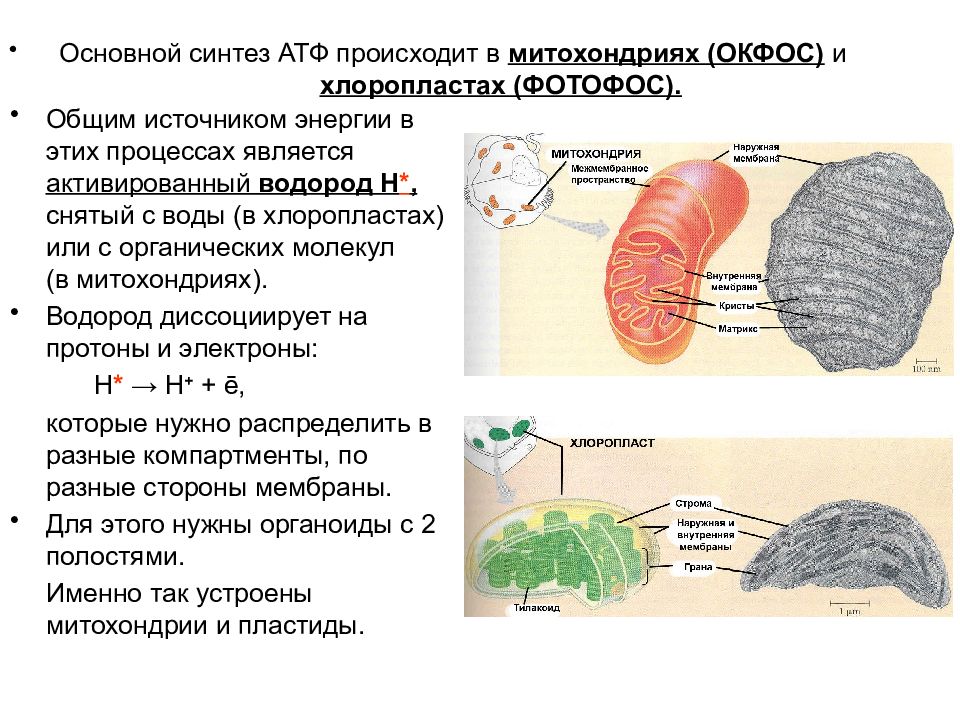 Цинк относится к микроэлементам
Цинк относится к микроэлементам
Б. В состав хлорофилла входит железо.
№13
Верны ли следующие суждения о строении и функциях органоидов?
А. Лизосомы относятся у двумембранным органоидам.
Б. У кишечной палочки АТФ синтезируются не в митохондриях.№14
Верны ли следующие суждения об обмене веществ?
А. Второй этап катаболизма – неполное окисление или бескислородный – протекает в цитоплазме..
Б. В результате гликолиза из одной молекулы глюкозы образуется 2 молекулы АТФ.
№15
Верны ли следующие суждения об особенностях ферментов?
А. Ферменты – это специфические белки, которые присутствуют во всех живых организмах и играют роль биологических катализаторов.
Б. Ферменты в отличие от химических катализаторов способны катализировать несколько различных реакций.
№16
Верны ли следующие суждения о приспособленности организмов?
Б. Японские макаки зимой при наступлении холодов спускаются с гор к термальным источникам и подолгу греются в теплой воде – это пример физиологической адаптации.
№17
Верны ли следующие суждения о происхождении жизни?
А. Невозможность самозарождения была доказана опытами Ф. Реди, Л. Пастера и др.
Б. Доказательствами инопланетного происхождения жизни служат НЛО, наскальные изображения летательных аппаратов
№18
А. Молекулы ДНК, находящиеся в митохондриях и хлоропластах, не являются хранителями наследственной информации.
№19
А. Плазматическая мембрана состоит из лидидного бислоя и встроенных белков.
Б. Мембранные белки выполняют транспортные, рецепторные и другие функции.
№20
А. В разных фазах мейоза клетка может нести диплоидный или гаплоидный набор хромосом.
Б. В течении всего митоза клетка кожи человека всегда диплоидна.
Верны ли следующие суждения:
№1 Верно – Б
№2: Верны обе формулировки
№3: Верно только А (борьба за существование и мутации – это факторы эволюции, а не результат)
№4 верно А (Б – неверно, т.к. сохранению в популяции наиболее приспособленных особей способствует естественный отбор, а наследственная изменчивость представляет собой материал для отбора).
№5 верны оба суждения
№6 Верно Б (А – неверно, т.к. упрощение в строении животных, связанное с паразитическим образом жизни, относят к дегенерации. А дегенерация, как и ароморфоз и идиоадаптация, тоже относится к биологическому процессу.
№7 Оба суждения неверны. А – генетический под триптолетен, т.е. в молекуле НК одна аминокислота кодируется сочетанием 3-х последовательно расположенных нуклеотидов.
Б – генетический код однозначен, т.е. один триплет (кодон) всегда кодирует только одну А.К.
№8 Верно только Б.
№9 Верно А
№10 Верны оба суждения
№11 Верно А
№12 Верно А
№13 Верно Б
№14 Верны оба суждения
№15 Верно А
№16 Неверны оба суждения
№17 Верно А
№18 Верно Б
№19 Верны А и Б
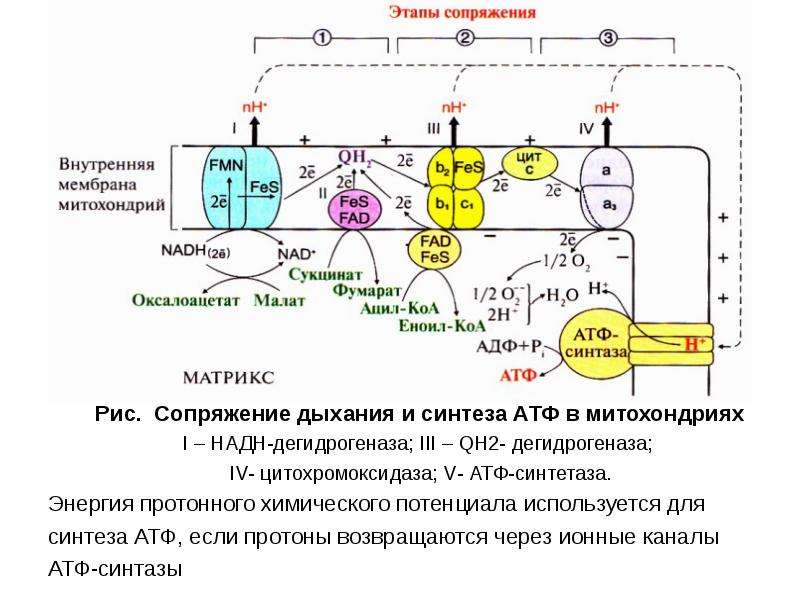
Митохондрии помогут диагностировать наследственные заболевания
Группа исследователей из Московского государственного университета им. М.В. Ломоносова в рамках программы развития МГУ и сотрудничества с Российским научным фондом разработала уникальную методику, благодаря которой в биологической практике впервые появилась возможность селективно исследовать перенос электронов между белковыми комплексами в живой митохондрии неразрушающими методами анализа, сообщает пресс-служба МГУ им. М.В. Ломоносова. Подробно о своей работе ученые рассказали в статье, опубликованной в последнем выпуске быстро набирающего популярность журнала Scientific Reports.
М.В. Ломоносова в рамках программы развития МГУ и сотрудничества с Российским научным фондом разработала уникальную методику, благодаря которой в биологической практике впервые появилась возможность селективно исследовать перенос электронов между белковыми комплексами в живой митохондрии неразрушающими методами анализа, сообщает пресс-служба МГУ им. М.В. Ломоносова. Подробно о своей работе ученые рассказали в статье, опубликованной в последнем выпуске быстро набирающего популярность журнала Scientific Reports.
Митохондрия – важнейшая внутриклеточная органелла, которую часто называют энергетической станцией клетки. В ней синтезируется АТФ – универсальное «топливо» организма, а ключевую роль в этом энергоснабжении играет перенос электронов между специальными белковыми комплексами, важнейший из которых носит название цитохром С.
 С этой точки зрения митохондрии крайне интересны и генетикам разных направлений и, главное, медикам, которые заняты изучением наследственных, как правило, трудноизлечимых заболеваний.
С этой точки зрения митохондрии крайне интересны и генетикам разных направлений и, главное, медикам, которые заняты изучением наследственных, как правило, трудноизлечимых заболеваний. По словам одного из основных авторов статьи, Надежды Браже, кандидата биологических наук с кафедры биофизики биологического факультета МГУ, сегодня в мире разработано множество методов исследования митохондрий, однако даже самые продвинутые и хитроумные из них не дают полного представления о том, что происходит между митохондриальными мембранами и с самим цитохромом С при переносе электронов.
Метод гигантской или поверхностно-усиленной рамановской спектроскопии
Очень перспективным в этом смысле представляется метод так называемой гигантской или поверхностно-усиленной рамановской спектроскопии (англ. SERS — surface-enhanced Raman Spectroscopy или спектроскопия гигантского комбинационного рассеяния), который и применили ученые из МГУ. Этот метод уже давно и активно используется в разного рода физических и химических экспериментах.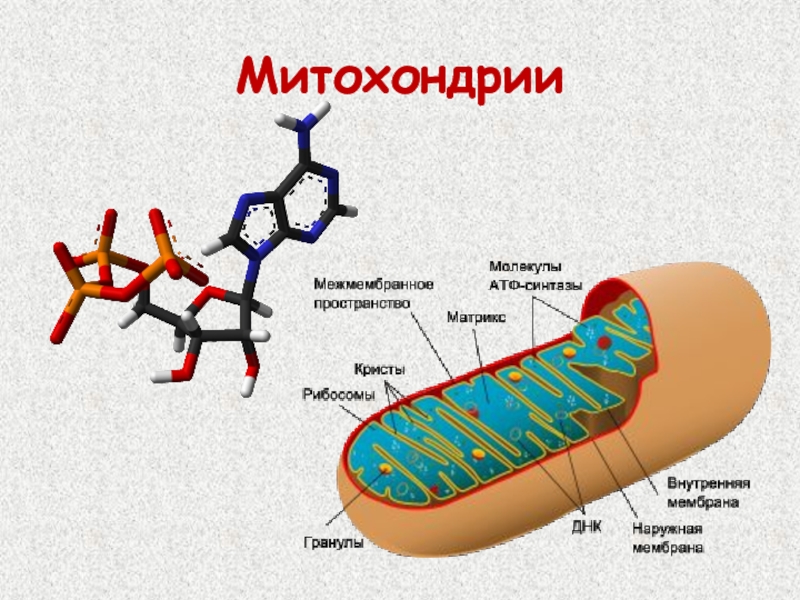 Где-то с середины-конца 1990-х годов XX века этим методом заинтересовались биологи и медики, занимающиеся изучением свойств и строением молекул в составе живых клеток.
Где-то с середины-конца 1990-х годов XX века этим методом заинтересовались биологи и медики, занимающиеся изучением свойств и строением молекул в составе живых клеток.
Существует, как известно, два вида рассеяния – релеевское, или упругое, при котором фотон отскакивает от препятствия, как мячик, не меняя своей частоты, и рамановское (комбинационное), или неупругое, при котором фотон при соударении с молекулой меняет ее энергетическое состояние. В результате этого он меняет и свою частоту и уносит в пространство некую толику информации о строении ударенной им молекулы, информации, которую в принципе можно извлечь для дальнейшего изучения.

Проблема разрешилась, когда в 1974 году был открыт эффект, при котором рамановские фотоны, рассеянные исследуемым объектом, находящимся вблизи наноструктурированной металлической поверхности, чудесным образом «усиливались» в миллиарды раз. До конца причины этого явления не выяснены, но считается, что здесь работают плазмоны — квазичастицы, представляющие собой осцилляции поверхностных электронов относительно положительно заряженных ядер металла наноструктуры. При совпадении собственной частоты плазмона и рамановского фотона возникает резонанс, который и позволяет сделать видимым почти невидимое. На основе этого эффекта вскоре появилась и соответствующая спектроскопия – SERS.
Биологи, занимающиеся исследованиями свойств биомакромолекул, быстро поняли, что такая спектроскопия может стать многообещающим способом извлекать информацию о структуре и работе молекул внутри живых органелл или клеток, не разрушая их. И с середины нулевых годов этого века начали довольно активно пытаться эту методику использовать, однако до сих пор эти попытки особых успехов не приносили.
Причин для неуспеха было более чем достаточно, но главные, по словам Надежды Браже, имели отношение к наноповерхностям, приводящим к гигантскому рамановскому рассеянию: из тысяч появившихся к тому времени вариантов одни не подходили для создания резонанса на нужных частотах, другие оказывались для митохондрий токсичными или сами быстро деградировали при помещении в физиологические жидкости.
«Ключевым моментом нашего достижения стал междисциплинарный подход к работе, в которую были вовлечены биологи, химики и физики. Это и привело к созданию уникальных наноструктурированных поверхностей и нового методического подхода в изучении митохондрий. Успех был бы невозможен без наших коллег с факультета наук о материалах МГУ, – приводит пресс-служба вуза слова кандидата биологических наук с кафедры биофизики биологического факультета МГУ Надежды Браже. – Молодые сотрудницы и магистрантки факультета после долгих и тщательных поисков нашли нужную и нетоксичную наноструктуру, что и позволило нам успешно провести работу».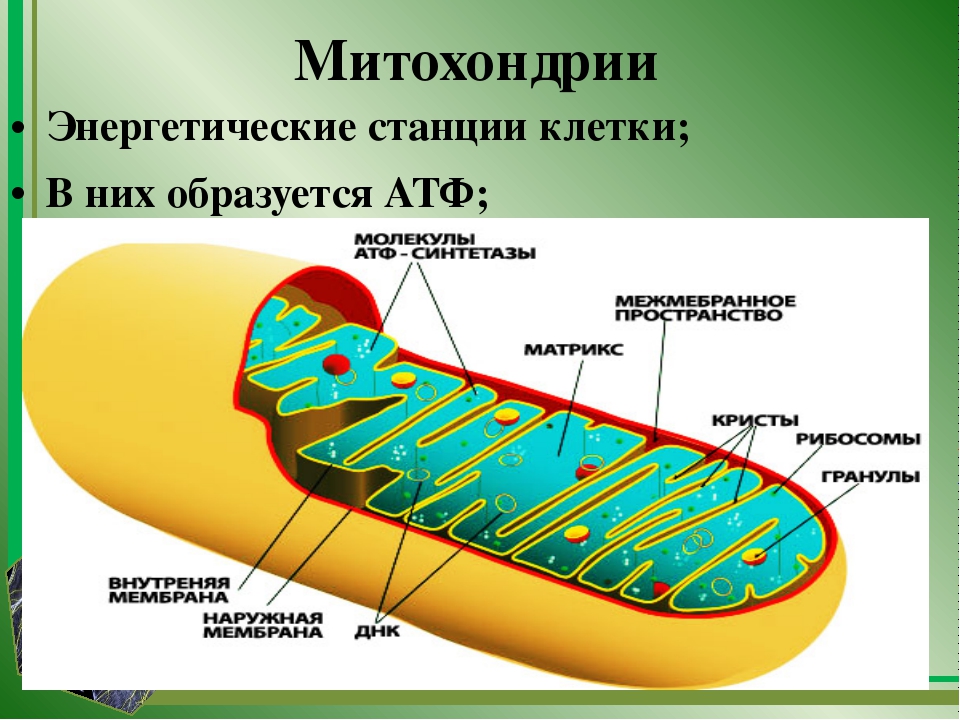
Поиски путей решения проблемы, как с точки зрения биологии, так и нанохимии и материаловедения на обоих факультетах действительно были долгими, они длились несколько лет и простотой вовсе не отличались. Порой тот или иной успех казался исследователям чудом, правда, обыкновенным чудом, которое можно научно обосновать, после чего из тысяч выбрать один-единственный вариант.
В результате после всех этих сложностей все получилось на удивление просто: на серебряную наноструктурированную поверхность уникальной наноструктуры помещали разбавленную суспензию митохондрий, облучали ее слабым лазерным пучком, а далее анализировали полученные спектры SERS. Оказалось, что при такой постановке эксперимента происходило многократное усиление комбинационного рассеяния только от молекул цитохрома С, благодаря чему можно было детально исследовать изменения, происходящие в его структуре при переносе электронов и синтезе АТФ. С помощью различных веществ транспорт электронов и синтез АТФ в митохондриях то запускали, то останавливали, что хорошо отражалось в регистрируемых спектрах.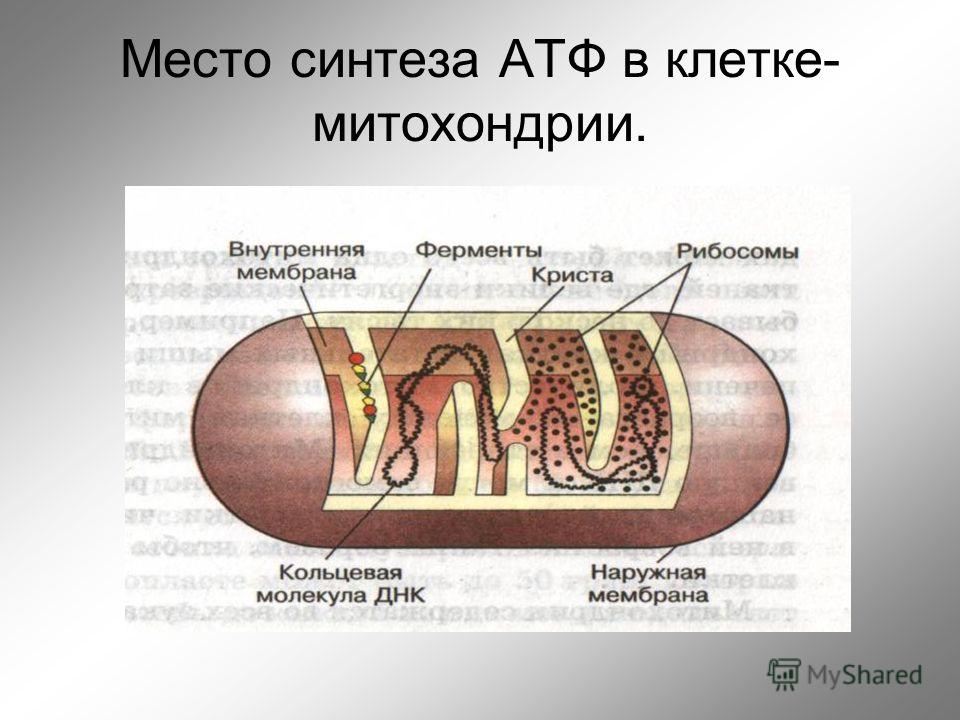
В ходе описанного исследования никаких эпохальных открытий вроде бы сделано не было. Однако фундаментально было сделано другое – получен новый метод, позволяющий эти открытия совершать. Этот метод предложен городу и миру, но Браже с коллегами тоже намерены воспользоваться своим творением.
«В качестве следующего шага хотим заняться исследованием митохондрий, выделенных из сердечных и скелетных мышц у крыс при заболеваниях сердечно-сосудистой системы и сахарном диабете, – приводит пресс-служба вуза слова Надежды Браже. – Мы надеемся, что полученные результаты позволят в дальнейшем разработать методику диагностики патологий на начальном этапе развития болезни, что позволит начинать лечение заболеваний раньше и эффективнее».
Страница не найдена |
Страница не найдена |
404. Страница не найдена
Страница не найдена
Архив за месяц
ПнВтСрЧтПтСбВс
2728293031
12
12
1
3031
12
15161718192021
25262728293031
123
45678910
12
17181920212223
31
2728293031
1
1234
567891011
12
891011121314
11121314151617
28293031
1234
12
12345
6789101112
567891011
12131415161718
19202122232425
3456789
17181920212223
24252627282930
12345
13141516171819
20212223242526
2728293031
15161718192021
22232425262728
2930
Архивы
Метки
Настройки
для слабовидящих
Митохондрия — это.
 .. Что такое Митохондрия?
Электронномикроскопическая фотография, показывающая митохондрии млекопитающего в поперечном сечении
.. Что такое Митохондрия?
Электронномикроскопическая фотография, показывающая митохондрии млекопитающего в поперечном сечении
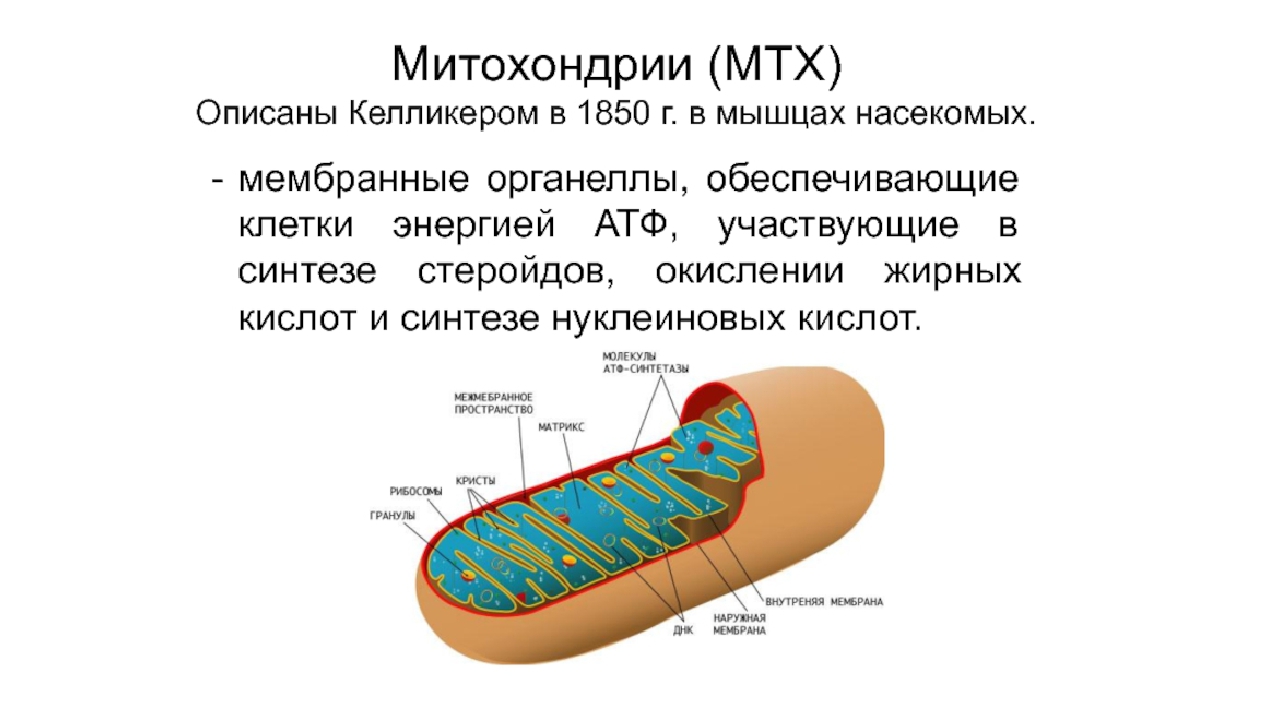
Митохо́ндрия (от греч. μίτος — нить и χόνδρος — зёрнышко, крупинка) — двумембранная гранулярная или нитевидная органелла толщиной около 0,5 мкм. Характерна для большинства эукариотических клеток как автотрофов (фотосинтезирующие растения), так и гетеротрофов (грибы, животные). Энергетическая станция клетки; основная функция — окисление органических соединений и использование освобождающейся при их распаде энергии в синтезе молекул АТФ, который происходит за счёт движения электрона по электронно-транспортной цепи белков внутренней мембраны. Количество митохондрий в клетках различных организмов существенно отличается: так, одноклеточные зелёные водоросли (эвглена, хлорелла, политомелла) и трипаносомы имеют лишь одну гигантскую митохондрию, тогда как ооцит и амёба Chaos chaos содержат 300 000 и 500 000 митохондрий соответственно; у кишечных анаэробных энтамёб и некоторых других паразитических простейших митохондрии отсутствуют.
Происхождение митохондрий
В соответствии с теорией симбиогенеза, митохондрии появились в результате захвата примитивными клетками (прокариотами) бактерий. Клетки, которые не могли сами использовать кислород для генерации энергии, имели серьёзные ограничения в возможностях развития; бактерии же (прогеноты) могли это делать. В процессе развития таких отношений прогеноты передали множество своих генов сформировавшемуся, благодаря повысившейся энергоэффективности, ядру теперь уже эукариот.[1] Вот почему современные митохондрии больше не являются самостоятельными организмами. Хотя их геном кодирует компоненты собственной системы синтеза белка, многие ферменты и белки, необходимые для их функции, кодируются хромосомами, синтезируются в клетке и только потом транспортируются в органеллы.
Митохондрии в клетке
Впервые митохондрии обнаружены в виде гранул в мышечных клетках в 1850 году. Число митохондрий в клетке непостоянно. Их особенно много в клетках, в которых потребность в кислороде велика. По своему строению они представляют собой цилиндрические органеллы, встречающиеся в эукариотической клетке в количестве от нескольких сот до 1-2 тысяч и занимающие 10-20 % её внутреннего объёма. Сильно варьируют так же размеры (от 1 до 70 мкм) и форма митохондрий. При этом ширина этих органелл относительно постоянна (0,5-1 мкм). Способны изменять форму. В зависимости от того, в каких участках клетки в каждый конкретный момент происходит повышенное потребление энергии, митохондрии способны перемещаться по цитоплазме в зоны наибольшего энергопотребления, используя для движения структуры цитоскелета эукариотической клетки.
По своему строению они представляют собой цилиндрические органеллы, встречающиеся в эукариотической клетке в количестве от нескольких сот до 1-2 тысяч и занимающие 10-20 % её внутреннего объёма. Сильно варьируют так же размеры (от 1 до 70 мкм) и форма митохондрий. При этом ширина этих органелл относительно постоянна (0,5-1 мкм). Способны изменять форму. В зависимости от того, в каких участках клетки в каждый конкретный момент происходит повышенное потребление энергии, митохондрии способны перемещаться по цитоплазме в зоны наибольшего энергопотребления, используя для движения структуры цитоскелета эукариотической клетки.
Альтернативой множеству разрозненных небольших митохондрий, функционирующих независимо друг от друга и снабжающих АТФ небольшие участки цитоплазмы, является существование длинных и разветвлённых митохондрий, каждая из которых может энергетически обеспечивать отдалённые друг от друга участки клетки (например, у одноклеточных зелёных водорослей Chlorella). Вариантом такой протяжённой системы может также являться упорядоченное пространственное объединение множества митохондрий (хондриом или митохондрион), обеспечивающее их кооперативную работу и встречающееся как у одноклеточных, так и у многоклеточных организмов. Особенно сложно этот тип хондриома устроен в скелетных мышцах млекопитающих, где группы гигантских разветвлённых митохондрий связаны друг с другом с помощью межмитохондриальных контактов (ММК). Последние образованы плотно прилегающими друг к другу наружными митохондриальными мембранами, в результате чего межмембранное пространство в этой зоне имеет повышенную электронную плотность. Особенно обильно ММК представлены в клетках сердечных мышц, где они связывают множественные отдельные митохондрии в согласованную работающую кооперативную систему.
Особенно сложно этот тип хондриома устроен в скелетных мышцах млекопитающих, где группы гигантских разветвлённых митохондрий связаны друг с другом с помощью межмитохондриальных контактов (ММК). Последние образованы плотно прилегающими друг к другу наружными митохондриальными мембранами, в результате чего межмембранное пространство в этой зоне имеет повышенную электронную плотность. Особенно обильно ММК представлены в клетках сердечных мышц, где они связывают множественные отдельные митохондрии в согласованную работающую кооперативную систему.
Структура митохондрий
Схема строения митохондрииНаружная мембрана
Наружная мембрана митохондрии имеет толщину около 7 нм, не образует впячиваний и складок, и замкнута сама на себя. На наружную мембрану приходится около 7 % от площади поверхности всех мембран клеточных органелл. Основная функция — отграничение митохондрии от цитоплазмы. Наружная мембрана митохондрии состоит из билипидного слоя и пронизывающих его белков; соотношение липидов и белков по массе — примерно 1:1. Особую роль играет порин — каналообразующий белок: он формирует в наружной мембране отверстия диаметром 2-3 нм, через которые могут проникать небольшие молекулы и ионы весом до 5 кДа. Крупные молекулы могут пересекать наружную мембрану только посредством активного транспорта через транспортные белки митохондриальных мембран. Для наружной мембраны характерно присутствие ферментов: монооксигеназы, ацил-СоА-синтетазы и фосфолипазы А2. Наружная мембрана митохондрии может взаимодействовать с мембраной эндоплазматического ретикулума; это играет важную роль в транспортировке липидов и ионов кальция.
Особую роль играет порин — каналообразующий белок: он формирует в наружной мембране отверстия диаметром 2-3 нм, через которые могут проникать небольшие молекулы и ионы весом до 5 кДа. Крупные молекулы могут пересекать наружную мембрану только посредством активного транспорта через транспортные белки митохондриальных мембран. Для наружной мембраны характерно присутствие ферментов: монооксигеназы, ацил-СоА-синтетазы и фосфолипазы А2. Наружная мембрана митохондрии может взаимодействовать с мембраной эндоплазматического ретикулума; это играет важную роль в транспортировке липидов и ионов кальция.
Межмембранное пространство
Межмембранное пространство представляет собой пространство между наружной и внутренней мембранами митохондрии. Его толщина — 10-20 нм. Так как наружная мембрана митохондрии проницаема для небольших молекул и ионов, их концентрация в периплазматическом пространстве мало отличается от таковой в цитоплазме. Напротив, крупным белкам для транспорта из цитоплазмы в периплазматическое пространство необходимо иметь специфические сигнальные пептиды; поэтому белковые компоненты периплазматического пространства и цитоплазмы различны. Одним из белков, содержащихся в периплазматическом пространстве, является цитохром c — один из компонентов дыхательной цепи митохондрий.
Одним из белков, содержащихся в периплазматическом пространстве, является цитохром c — один из компонентов дыхательной цепи митохондрий.
Внутренняя мембрана
Внутренняя мембрана образует многочисленные гребневидные складки — кристы, существенно увеличивающие площадь ее поверхности и, например, в клетках печени составляет около трети всех клеточных мембран. Характерной чертой состава внутренней мембраны митохондрий является присутствие в ней кардиолипина — особого фосфолипида, содержащего сразу четыре жирные кислоты и делающего мембрану абсолютно непроницаемой для протонов. Ещё одна особенность внутренней мембраны митохондрий — очень высокое содержание белков (до 70 % по весу), представленных транспортными белками, ферментами дыхательной цепи, а также крупными АТФ-синтетазными комплексами. Внутренняя мембрана митохондрии в отличие от внешней не имеет специальных отверстий для транспорта мелких молекул и ионов; на ней, на стороне, обращенной к матриксу, располагаются особые молекулы АТФ-синтазы, состоящие из головки, ножки и основания.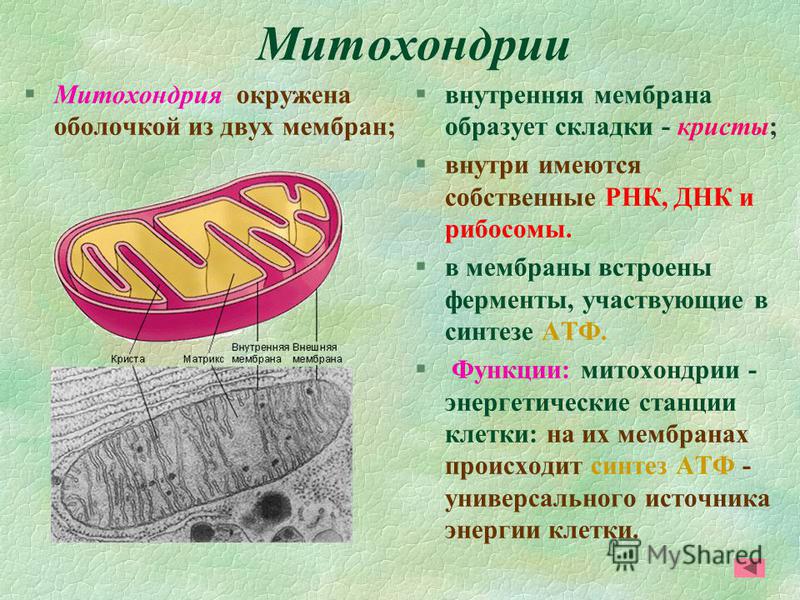 При прохождении через них протонов происходит синтез АТФ. В основании частиц, заполняя собой всю толщу мембраны, располагаются компоненты дыхательной цепи. Наружная и внутренняя мембраны в некоторых местах соприкасаются, там находится специальный белок-рецептор, способствующий транспорту митохондриальных белков, закодированных в ядре, в матрикс митохондрии.
При прохождении через них протонов происходит синтез АТФ. В основании частиц, заполняя собой всю толщу мембраны, располагаются компоненты дыхательной цепи. Наружная и внутренняя мембраны в некоторых местах соприкасаются, там находится специальный белок-рецептор, способствующий транспорту митохондриальных белков, закодированных в ядре, в матрикс митохондрии.
Матрикс
Матрикс — ограниченное внутренней мембраной пространство. В матриксе (розовом веществе) митохондрии находятся ферментные системы окисления пирувата, жирных кислот, а также ферменты цикла трикарбоновых кислот (цикла Кребса). Кроме того, здесь же находится митохондриальная ДНК, РНК и собственный белоксинтезирующий аппарат митохондрии.
Митохондриальная ДНК
Находящаяся в матриксе митохондриальная ДНК представляет собой замкнутую кольцевую двуспиральную молекулу, в клетках человека имеющую размер 16569 нуклеотидных пар, что приблизительно в 105 раз меньше ДНК, локализованной в ядре. В целом митохондриальная ДНК кодирует 2 рРНК, 22 тРНК и 13 субъединиц ферментов дыхательной цепи, что составляет не более половины обнаруживаемых в ней белков.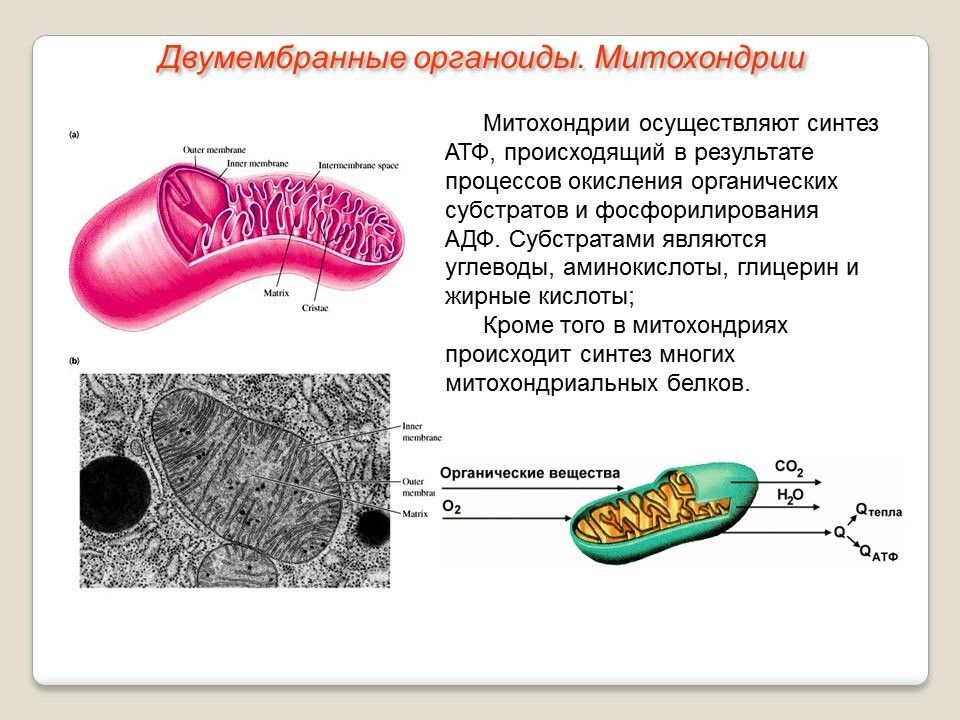 В частности, под контролем митохондрального генома кодируются семь субъединиц АТФ-синтетазы, три субъединицы цитохромоксидазы и одна субъединица убихинол-цитохром-с-редуктазы. При этом все белки, кроме одного, две рибосомные и шесть тРНК транскрибируются с более тяжёлой (наружной) цепи ДНК, а 14 других тРНК и один белок транскрибируются с более лёгкой (внутренней) цепи.
В частности, под контролем митохондрального генома кодируются семь субъединиц АТФ-синтетазы, три субъединицы цитохромоксидазы и одна субъединица убихинол-цитохром-с-редуктазы. При этом все белки, кроме одного, две рибосомные и шесть тРНК транскрибируются с более тяжёлой (наружной) цепи ДНК, а 14 других тРНК и один белок транскрибируются с более лёгкой (внутренней) цепи.
На этом фоне геном митохондрий растений значительно больше и может достигать 370000 нуклеотидных пар, что примерно в 20 раз больше описанного выше генома митохондрий человека. Количество генов здесь также примерно в 7 раз больше, что сопровождается появлением в митохондриях растений дополнительных путей электронного транспорта, не сопряжённых с синтезом АТФ.
Митохондриальная ДНК реплицируется в интерфазе, что частично синхронизировано с репликацией ДНК в ядре. Во время же клеточного цикла митохондрии делятся надвое путём перетяжки, образование которой начинается с кольцевой бороздки на внутренней митохондриальной мембране. Детальное изучение нуклеотидной последовательности митохондриального генома позволило установить то, что в митохондриях животных и грибов нередки отклонения от универсального генетического кода. Так, в митохондриях человека кодон ТАТ вместо изолейцина в стандартном коде кодирует аминокислоту метионин, кодоны ТСТ и ТСС, обычно кодирующие аргинин, являются стоп-кодонами, а кодон АСТ, в стандартном коде являющийся стоп-кодоном, кодирует аминокислоту метионин. Что касается митохондрий растений, то, по-видимому, они используют универсальный генетический код. Другой чертой митохондрий является особенность узнавания кодонов тРНК, заключающаяся в том, что одна подобная молекула способна узнавать не один, но сразу три или четыре кодона. Указанная особенность снижает значимость третьего нуклеотида в кодоне и приводит к тому, что митохондрии требуется меньшее разнообразие типов тРНК. При этом достаточным количеством оказываются всего 22 различных тРНК.
Детальное изучение нуклеотидной последовательности митохондриального генома позволило установить то, что в митохондриях животных и грибов нередки отклонения от универсального генетического кода. Так, в митохондриях человека кодон ТАТ вместо изолейцина в стандартном коде кодирует аминокислоту метионин, кодоны ТСТ и ТСС, обычно кодирующие аргинин, являются стоп-кодонами, а кодон АСТ, в стандартном коде являющийся стоп-кодоном, кодирует аминокислоту метионин. Что касается митохондрий растений, то, по-видимому, они используют универсальный генетический код. Другой чертой митохондрий является особенность узнавания кодонов тРНК, заключающаяся в том, что одна подобная молекула способна узнавать не один, но сразу три или четыре кодона. Указанная особенность снижает значимость третьего нуклеотида в кодоне и приводит к тому, что митохондрии требуется меньшее разнообразие типов тРНК. При этом достаточным количеством оказываются всего 22 различных тРНК.
Имея собственный генетический аппарат, митохондрия обладает и собственной белоксинтезирующей системой, особенностью которой в клетках животных и грибов являются очень маленькие рибосомы, характеризуемые коэффициентом седиментации 55S, что даже ниже аналогичного показателя у 70s-рибосом прокариотического типа. При этом две большие рибосомные РНК также имеют меньшие размеры, чем у прокариот, а малая рРНК вообще отсутствует. В митохондриях растений, напротив, рибосомы более сходны с прокариотическими по размерам и строению.
При этом две большие рибосомные РНК также имеют меньшие размеры, чем у прокариот, а малая рРНК вообще отсутствует. В митохондриях растений, напротив, рибосомы более сходны с прокариотическими по размерам и строению.
Митохондриальные белки
Количество транслируемых с митохондриальной мРНК белков, формирующих субъединицы крупных ферментных комплексов, ограниченно. Значительная часть белков кодируется в ядре и синтезируется на цитоплазматических 80S рибосомах. В частности, так образуются некоторые белки — переносчики электронов, митохондриальные транслоказы, компоненты транспорта белков в митохондрии, а также факторы, необходимые для транскрипции, трансляции и репликации митохондриальной ДНК. При этом подобные белки на своём N-конце имеют особые сигнальные пептиды, размер которых варьирует от 12 до 80 аминокислотных остатков. Данные участки формируют амфифильные завитки, обеспечивают специфический контакт белков со связывающими доменами митохондриальных распознающих рецепторов, локализованных на наружной мембране.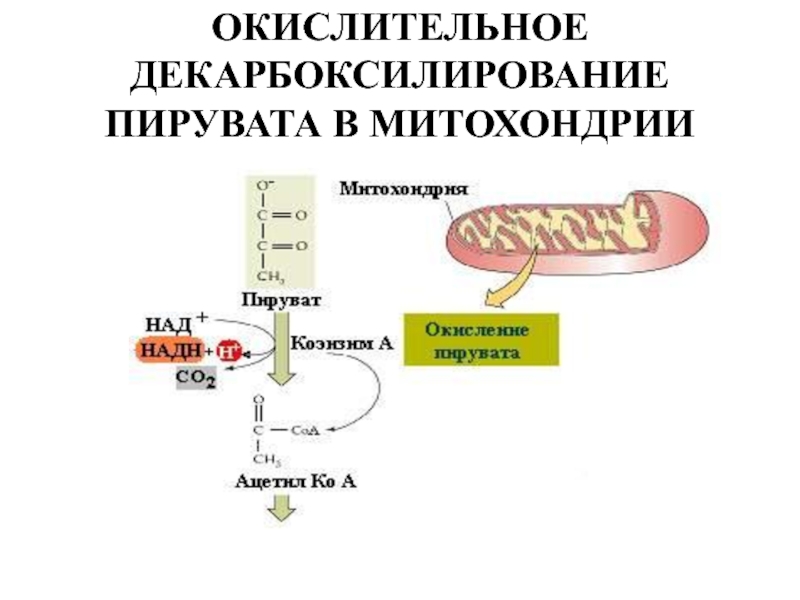 До наружной мембраны митохондрии данные белки транспортируются в частично развёрнутом состоянии в ассоциации с белками-шаперонами (в частности — с hsp70). После переноса через наружную и внутреннюю мембраны в местах их контактов поступающие в митохондрию белки вновь связываются с шаперонами, но уже собственного митохондриального происхождения, которые подхватывают пересекающий мембраны белок, способствуют его втягиванию в митохондрию, а также контролируют процесс правильного сворачивания полипептидной цепи. Большинство шаперонов обладает АТФазной активностью, в результате чего как транспорт белков в митохондрию, так и образование их функционально активных форм являются энергозависимыми процессами.
До наружной мембраны митохондрии данные белки транспортируются в частично развёрнутом состоянии в ассоциации с белками-шаперонами (в частности — с hsp70). После переноса через наружную и внутреннюю мембраны в местах их контактов поступающие в митохондрию белки вновь связываются с шаперонами, но уже собственного митохондриального происхождения, которые подхватывают пересекающий мембраны белок, способствуют его втягиванию в митохондрию, а также контролируют процесс правильного сворачивания полипептидной цепи. Большинство шаперонов обладает АТФазной активностью, в результате чего как транспорт белков в митохондрию, так и образование их функционально активных форм являются энергозависимыми процессами.
Функции митохондрий и энергообразование
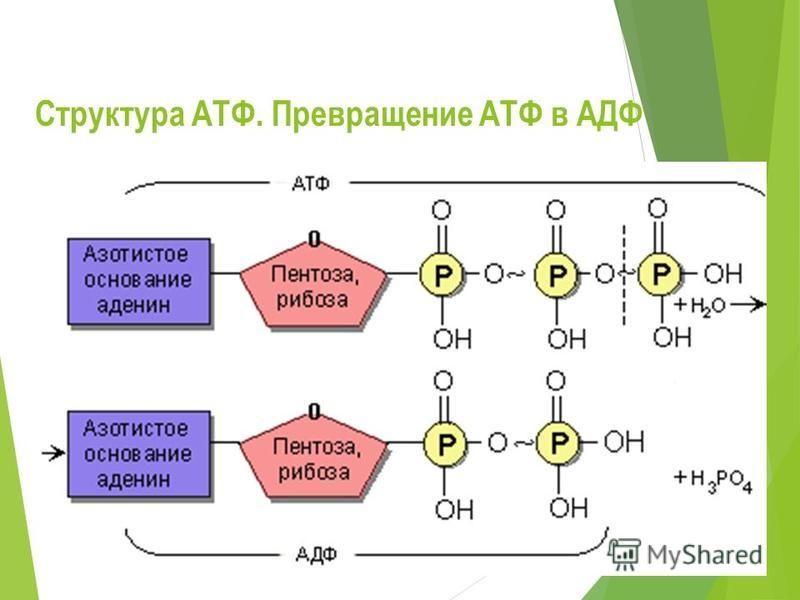
Основной функцией митохондрий является синтез АТФ — универсальной формы химической энергии в любой живой клетке. Как и у прокариот, данная молекула может образовываться двумя путями: в результате субстратного фосфорилирования в жидкой фазе (например, при гликолизе) или в процессе мембранного фосфорилирования, связанного с использованием энергии трансмембранного электрохимического градиента (англ.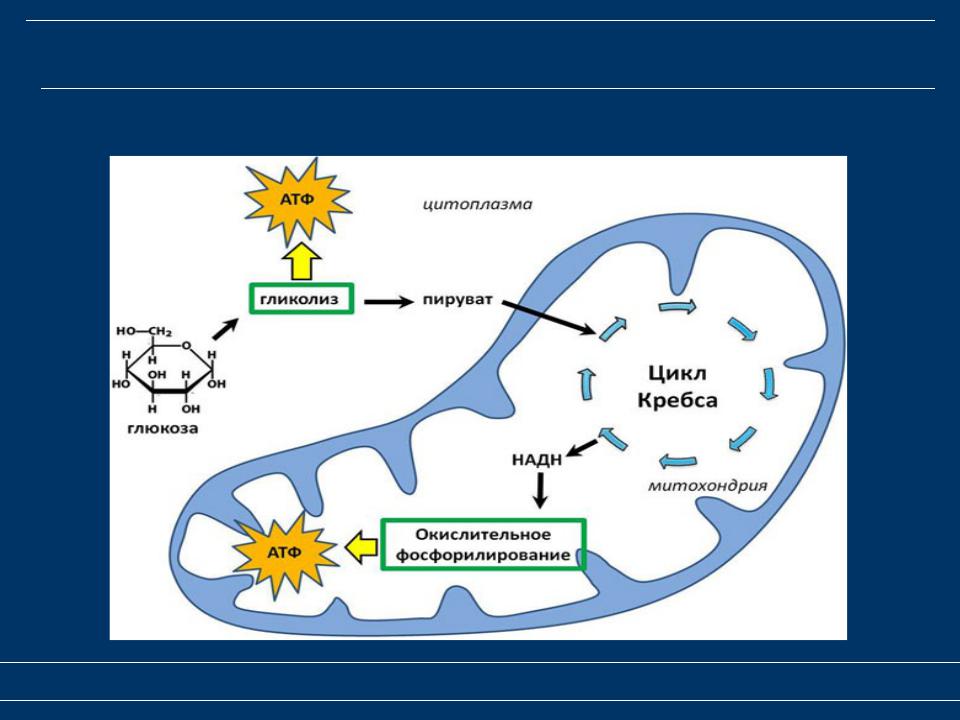 )русск. протонов (ионов водорода). Митохондрии реализуют оба эти пути, первый из которых характерен для начальных процессов окисления субстрата и происходит в матриксе, а второй завершает процессы энергообразования и связан с кристами митохондрий. При этом своеобразие митохондрий как энергообразующих органелл эукариотической клетки определяет именно второй путь генерации АТФ, получивший название «хемиосмотического сопряжения». По сути это последовательное превращение химической энергии восстанавливающих эквивалентов НАДН в электрохимический протонный градиент ΔμН+ по обе стороны внутренней мембраны митохондрии, что приводит в действие мембранно-связанную АТФ-синтетазу и завершается образованием макроэргической связи в молекуле АТФ.
)русск. протонов (ионов водорода). Митохондрии реализуют оба эти пути, первый из которых характерен для начальных процессов окисления субстрата и происходит в матриксе, а второй завершает процессы энергообразования и связан с кристами митохондрий. При этом своеобразие митохондрий как энергообразующих органелл эукариотической клетки определяет именно второй путь генерации АТФ, получивший название «хемиосмотического сопряжения». По сути это последовательное превращение химической энергии восстанавливающих эквивалентов НАДН в электрохимический протонный градиент ΔμН+ по обе стороны внутренней мембраны митохондрии, что приводит в действие мембранно-связанную АТФ-синтетазу и завершается образованием макроэргической связи в молекуле АТФ.
В целом весь процесс энергообразования в митохондриях может быть разбит на четыре основные стадии, первые две из которых протекают в матриксе, а две последние — на кристах митохондрий:
- Превращение поступивших из цитоплазмы в митохондрию пирувата и жирных кислот в ацетил-СоА;
- Окисление ацетил-СоА в цикле Кребса, ведущее к образованию НАДН;
- Перенос электронов с НАДН на кислород по дыхательной цепи;
- Образование АТФ в результате деятельности мембранного АТФ-синтетазного комплекса.
Ещё в цитоплазме в серии из 10 отдельных ферментативных реакций шестиуглеродная молекула глюкозы частично окисляется до двух трёхуглеродных молекул пирувата с образованием двух молекул АТФ. Затем пируват переносится из цитозоля через наружную и внутреннюю мембраны в матрикс, где первоначально превращается в ацетил-СоА. Этот процесс катализируется крупным пируватдегидрогеназным комплексом, имеющим размер, сопоставимый с размером рибосомы, и состоящим из трёх ферментов, пяти коферментов и двух регуляторных белков. Точно также жирные кислоты, полученные при расщеплении нерастворимых триглицеридов в цитоплазме, переносятся в митохондриальный матрикс в виде ацетил-СоА-производных.
На следующем этапе, также протекающем в матриксе митохондрии, ацетил-СоА полностью окисляется в цикле Кребса. В его работе задействованы четыре отдельных фермента, за каждый цикл обеспечивающие укорочение углеводородной цепи на два атома углерода, которые в дальнейшем превращаются в СО2.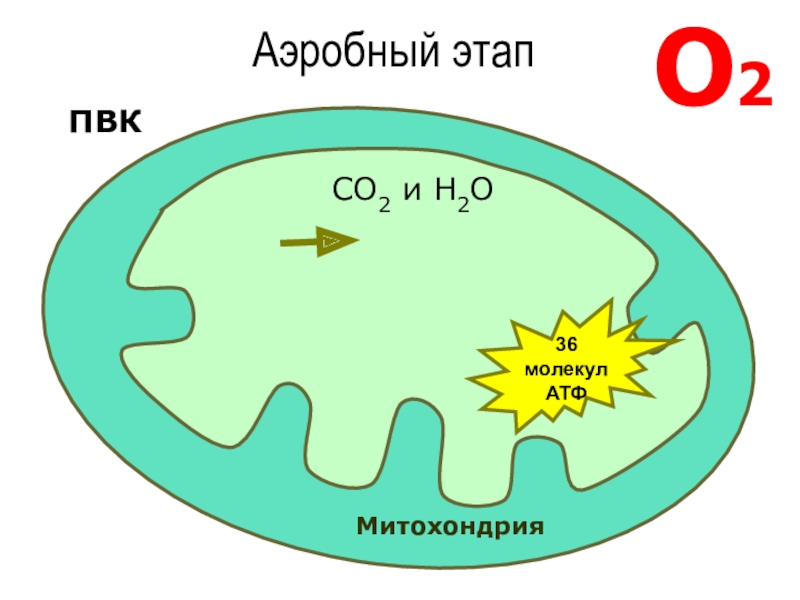 Этот процесс обеспечивает образование одной молекулы ГТФ, а также НАДН — высокоэнергетического промежуточного соединения, которое легко отдаёт электроны в цепь переноса электронов на кристах митохондрий.
Этот процесс обеспечивает образование одной молекулы ГТФ, а также НАДН — высокоэнергетического промежуточного соединения, которое легко отдаёт электроны в цепь переноса электронов на кристах митохондрий.
Дальнейшие процессы энергообразования в митохондрии происходят на её кристах и связаны с переносом электронов от НАДН к кислороду. В соответствии с тем, что потребление кислорода в качестве окислителя обычно называют «внутриклеточным дыханием», электронно-транспортную цепь ферментов, осуществляющих последовательный перенос электронов от НАДН к кислороду, часто называют «дыхательной цепью». При этом трансформация энергии окисления осуществляется ферментами, расположенными на кристах митохондрий и осуществляющими векторный (направленный по отношению к сторонам мембраны) перенос протонов водорода из матрикса митохондрии в межмембранное пространство. В этом состоит принципиальное отличие работы оксидоредуктаз дыхательной цепи от функционирования ферментов, катализирующих реакции в гомогенном (изотропном) растворе, где вопрос о направлении реакции в пространстве не имеет смысла.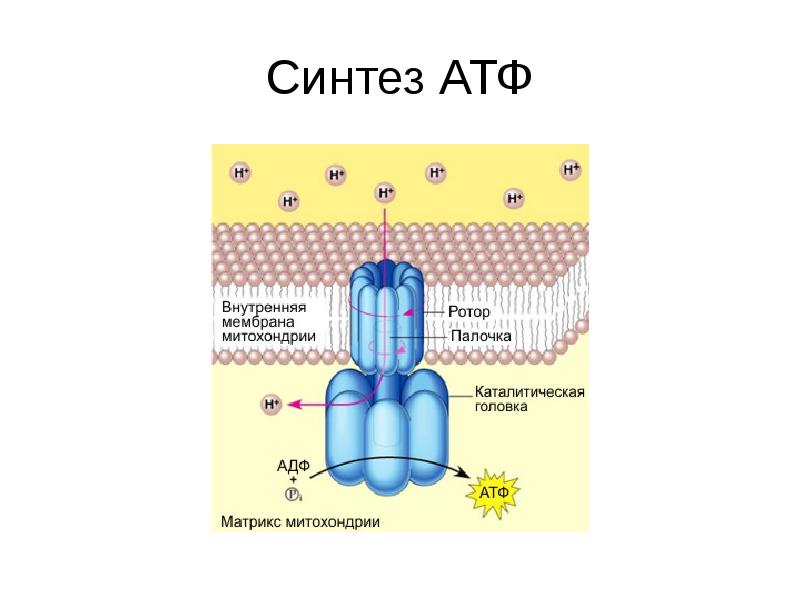
Весь процесс переноса электрона по дыхательной цепи может быть разбит на три стадии, каждая из которых катализируется отдельным трансмембранным липопротеидным комплексом (I, III и IV), встроенным в мембрану кристы митохондрии. В состав каждого из названных комплексов входят следующие компоненты:
- Большой олигомерный фермент, катализирующий перенос электронов;
- Небелковые органические (простетические) группы, принимающие и высвобождающие электроны;
- Белки, обеспечивающие движение электронов.
Каждый из этих комплексов осуществляет перенос электронов от донора к акцептору по градиенту редокс-потенциала через ряд последовательно функционирующих переносчиков. В качестве последних в дыхательной цепи митохондрий функционируют мигрирующие в плоскости мембраны жирорастворимые молекулы убихинона, а также небольшие (молекулярная масса 13 кДа) водорастворимые белки, содержащие ковалентно связанный гем и называемые «цитохромами с». При этом три из пяти компонентов, составляющих дыхательную цепь, работают так, что перенос электронов сопровождается переносом протонов через мембрану крист митохондрий в направлении из матрикса в межмембранное пространство.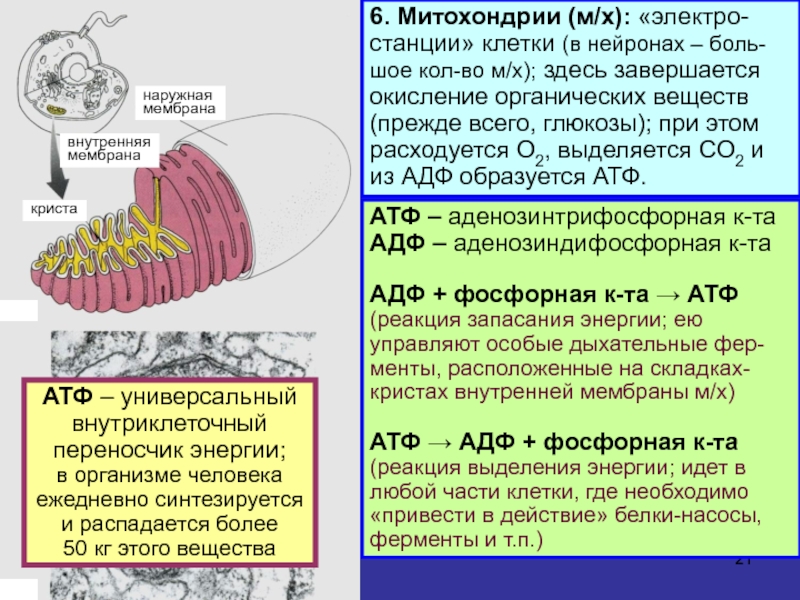
Дыхательная цепь начинается с комплекса I (НАДН-убихинон-оксидоредуктаза), состоящего из 16-26 полипептидных цепей и имеющего молекулярную массу около 850 кДа. Функциональная активность этого комплекса определяется тем, что он содержит в своём составе более 20 атомов железа, упакованных в ячейки из атомов серы, а также флавин (Фл — производное витамина рибофлавина). Комплекс I катализирует окисление НАДН, отщепляя от него два электрона, которые после «путешествия» по окислительно-восстановительным компонентам комплекса I попадают на молекулу-переносчик, в качестве которой выступает убихинон (Q). Последний способен ступенчато восстанавливаться, принимая на себя по два электрона и протона и, таким образом, превращаясь в восстановленную форму — убихинол (QH2).
Энергетический потенциал (запас энергии) в молекуле убихинола существенно ниже, чем в молекуле НАДН, а разница в подобной энергии временно запасается в виде особого вида — электрохимического протонного градиента. Последний возникает в результате того, что перенос электронов по простетическим группам комплекса I, ведущий к снижению энергетического потенциала электронов, сопровождается трансмембранным переносом двух протонов из матрикса в межмембранное пространство митохондрии.
Последний возникает в результате того, что перенос электронов по простетическим группам комплекса I, ведущий к снижению энергетического потенциала электронов, сопровождается трансмембранным переносом двух протонов из матрикса в межмембранное пространство митохондрии.
Восстановленный убихинол мигрирует в плоскости мембраны, где достигает второго фермента дыхательной цепи — комплекса III (bc1). Последний представляет собой димер из субъединиц b и c1 с молекулярной массой более 300 кДа, сформированный из восьми полипептидных цепей и содержащий атомы железа как в серных ячейках, так и в виде комплексов с гемами b(I), b(II) и c1 — сложными гетероциклическими молекулами с четырьмя атомами азота, расположенными по углам металлосвязывающего квадрата. Комплекс III катализирует реакцию восстановления убихинола до убихинона с передачей электронов на атом железа второй молекулы переносчика (находящегося в межмембранном пространстве цитохрома c). Отщепляющиеся при этом от убихинола два протона водорода освобождаются в межмембранное пространство, продолжая формирование электрохимического градиента. Наконец, ещё два протона водорода переносятся в межмембранное пространство митохондрии за счёт энергии электронов, проходящих по простетических группам комплекса III.
Отщепляющиеся при этом от убихинола два протона водорода освобождаются в межмембранное пространство, продолжая формирование электрохимического градиента. Наконец, ещё два протона водорода переносятся в межмембранное пространство митохондрии за счёт энергии электронов, проходящих по простетических группам комплекса III.
Последняя стадия катализируется комплексом IV (цитохром c-оксидаза) с молекулярной массой около 200 кДа, состоящим из 10-13 полипептидных цепей и, помимо двух различных гемов, включающим также несколько атомов меди, прочно связанных с белками. При этом электроны, отбираемые у восстановленного цитохрома c, пройдя по атомам железа и меди в составе комплекса IV, попадают на связанный в активном центре этого фермента кислород, что приводит к образованию воды.
Таким образом, суммарная реакция, катализируемая ферментами дыхательной цепи, состоит в окислении НАДН кислородом с образованием воды. По сути этот процесс заключается в ступенчатом переносе электронов между атомами металлов, присутствующих в простетических группах белковых комплексов дыхательной цепи, где каждый последующий комплекс обладает более высоким сродством к электрону, чем предыдущий.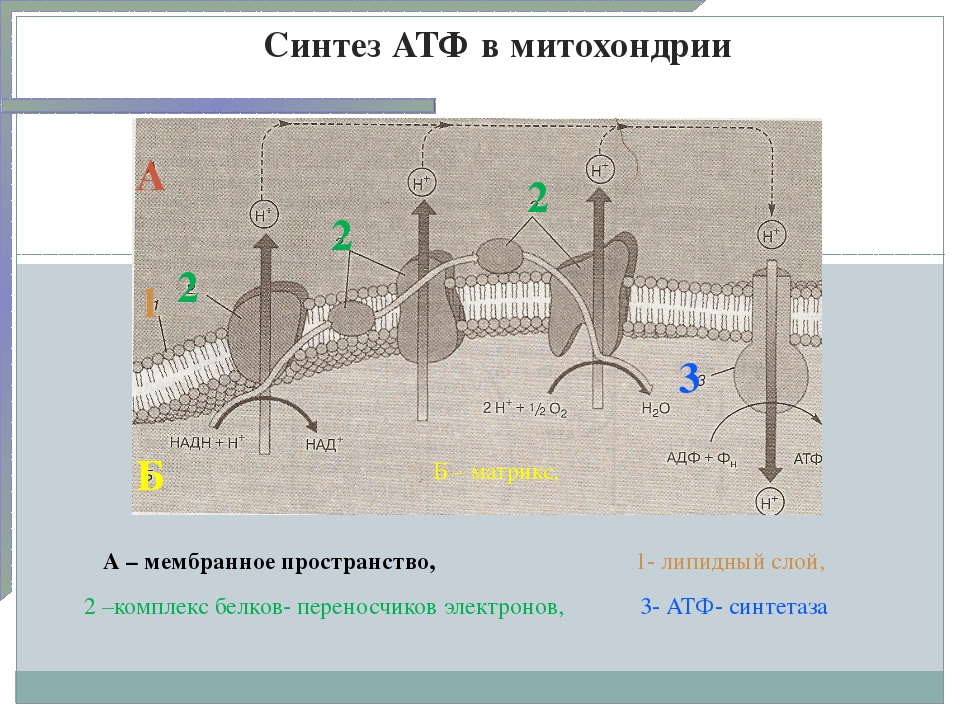 При этом сами электроны передаются по цепи до тех пор, пока не соединятся с молекулярным кислородом, обладающим наибольшим сродством к электронам. Освобождаемая же при этом энергия запасается в виде электрохимического (протонного) градиента по обе стороны внутренней мембраны митохондрий. При этом считается, что в процессе транспорта по дыхательной цепи пары электронов перекачивается от трёх до шести протонов.
При этом сами электроны передаются по цепи до тех пор, пока не соединятся с молекулярным кислородом, обладающим наибольшим сродством к электронам. Освобождаемая же при этом энергия запасается в виде электрохимического (протонного) градиента по обе стороны внутренней мембраны митохондрий. При этом считается, что в процессе транспорта по дыхательной цепи пары электронов перекачивается от трёх до шести протонов.
Завершающим этапом функционирования митохондрии является генерация АТФ, осуществляемая встроенным во внутреннюю мембрану специальным макромолекулярным комплексом с молекулярной массой 500 кДа. Этот комплекс, называемый АТФ-синтетазой, как раз и катализирует синтез АТФ путём конверсии энергии трансмембранного электрохимического градиента протонов водорода в энергию макроэргической связи молекулы АТФ.
АТФ-синтеза
В структурно-функциональном плане АТФ-синтаза состоит из двух крупных фрагментов, обозначаемых символами F1 и F0. Первый из них (фактор сопряжения F1) обращён в сторону матрикса митохондрии и заметно выступает из мембраны в виде сферического образования высотой 8 нм и шириной 10 нм.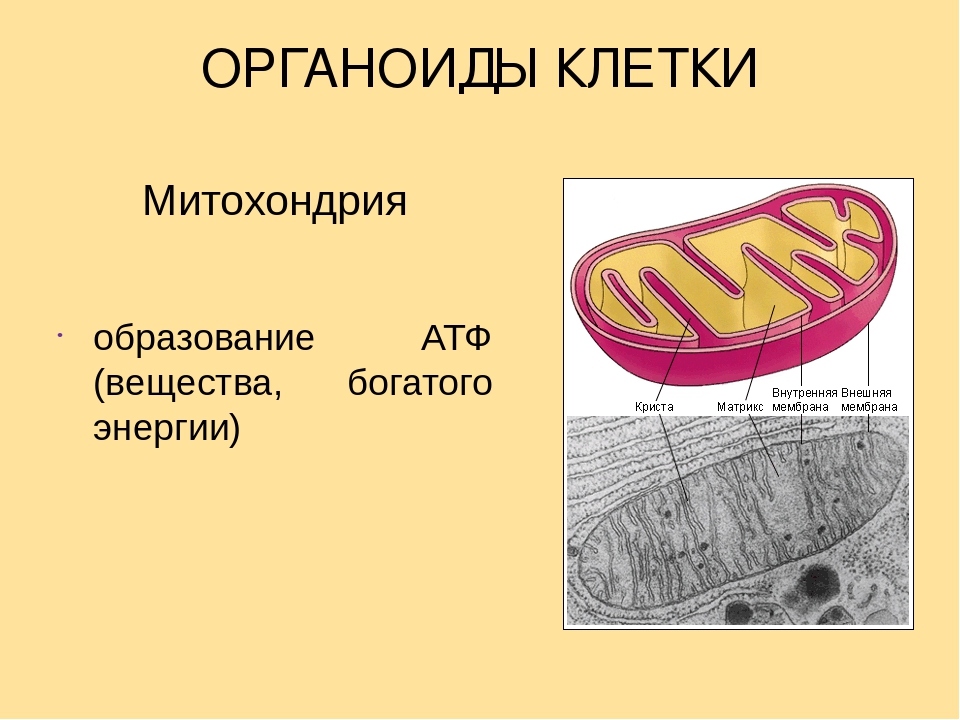 Он состоит из девяти субъединиц, представленных пятью типами белков. Полипептидные цепи трёх субъединиц α и стольких же субъединиц β уложены в похожие по строению белковые глобулы, которые вместе образуют гексамер (αβ)3, имеющий вид слегка приплюснутого шара. Подобно плотно уложенным долькам апельсина, последовательно расположенные субъединицы α и β образуют структуру, характеризующуюся осью симметрии третьего порядка с углом поворота 120°. В центре этого гексамера находится субъединица γ, которая образована двумя протяжёнными полипептидными цепями и напоминает слегка деформированный изогнутый стержень длиной около 9 нм. При этом нижняя часть субъединицы γ выступает из шара на 3 нм в сторону мембранного комплекса F0. Также внутри гексамера находится минорная субъединица ε, связанная с γ. Последняя (девятая) субъединица обозначается символом δ и расположена на внешней стороне F1.
Он состоит из девяти субъединиц, представленных пятью типами белков. Полипептидные цепи трёх субъединиц α и стольких же субъединиц β уложены в похожие по строению белковые глобулы, которые вместе образуют гексамер (αβ)3, имеющий вид слегка приплюснутого шара. Подобно плотно уложенным долькам апельсина, последовательно расположенные субъединицы α и β образуют структуру, характеризующуюся осью симметрии третьего порядка с углом поворота 120°. В центре этого гексамера находится субъединица γ, которая образована двумя протяжёнными полипептидными цепями и напоминает слегка деформированный изогнутый стержень длиной около 9 нм. При этом нижняя часть субъединицы γ выступает из шара на 3 нм в сторону мембранного комплекса F0. Также внутри гексамера находится минорная субъединица ε, связанная с γ. Последняя (девятая) субъединица обозначается символом δ и расположена на внешней стороне F1.
Мембранная часть АТФ-синтазы, называемая фактором сопряжения F0, представляет собой гидрофобный белковый комплекс, пронизывающий мембрану насквозь и имеющий внутри себя два полуканала для прохождения протонов водорода. Всего в состав комплекса F0 входит одна белковая субъединица типа а, две копии субъединицы b, а также от 9 до 12 копий мелкой субъединицы c. Субъединица а (молекулярная масса 20 кДа) полностью погружена в мембрану, где образует шесть пересекающих её α-спиральных участков. Субъединица b (молекулярная масса 30 кДа) содержит лишь один сравнительно короткий погружённый в мембрану α-спиральный участок, а остальная её часть заметно выступает из мембраны в сторону F1 и закрепляется за расположенную на её поверхности субъединицу δ. Каждая из 9-12 копий субъединицы c (молекулярная масса 6-11 кДа) представляет собой сравнительно небольшой белок из двух гидрофобных α-спиралей, соединённых друг с другом короткой гидрофильной петлёй, ориентированной в сторону F1, а все вместе образуют единый ансамбль, имеющий форму погружённого в мембрану цилиндра. Выступающая из комплекса F1 в сторону F0 субъединица γ как раз и погружена внутрь этого цилиндра и достаточно прочно зацеплена за него.
Всего в состав комплекса F0 входит одна белковая субъединица типа а, две копии субъединицы b, а также от 9 до 12 копий мелкой субъединицы c. Субъединица а (молекулярная масса 20 кДа) полностью погружена в мембрану, где образует шесть пересекающих её α-спиральных участков. Субъединица b (молекулярная масса 30 кДа) содержит лишь один сравнительно короткий погружённый в мембрану α-спиральный участок, а остальная её часть заметно выступает из мембраны в сторону F1 и закрепляется за расположенную на её поверхности субъединицу δ. Каждая из 9-12 копий субъединицы c (молекулярная масса 6-11 кДа) представляет собой сравнительно небольшой белок из двух гидрофобных α-спиралей, соединённых друг с другом короткой гидрофильной петлёй, ориентированной в сторону F1, а все вместе образуют единый ансамбль, имеющий форму погружённого в мембрану цилиндра. Выступающая из комплекса F1 в сторону F0 субъединица γ как раз и погружена внутрь этого цилиндра и достаточно прочно зацеплена за него.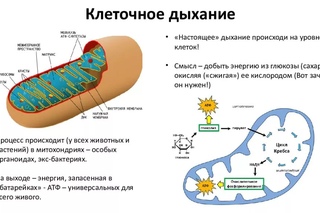
Таким образом, в молекуле АТФ-синтазы можно выделить две группы белковых субъединиц, которые могут быть уподоблены двум деталям мотора: ротору и статору. «Статор» неподвижен относительно мембраны и включает в себя шарообразный гексамер (αβ)3, находящуюся на его поверхности и субъединицу δ, а также субъединицы a и b мембранного комплекса F0. Подвижный относительно этой конструкции «ротор» состоит из субъединиц γ и ε, которые, заметно выступая из комплекса (αβ)3, соединяются с погружённым в мембрану кольцом из субъединиц c.
Способность синтезировать АТФ — свойство единого комплекса F0F1, сопряжённого с переносом протонов водорода через F0 к F1, в последнем из которых как раз и расположены каталитические центры, осуществляющие преобразование АДФ и фосфата в молекулу АТФ. Движущей же силой для работы АТФ-синтазы является протонный потенциал, создаваемый на внутренней мембране митохондрий в результате работы цепи электронного транспорта.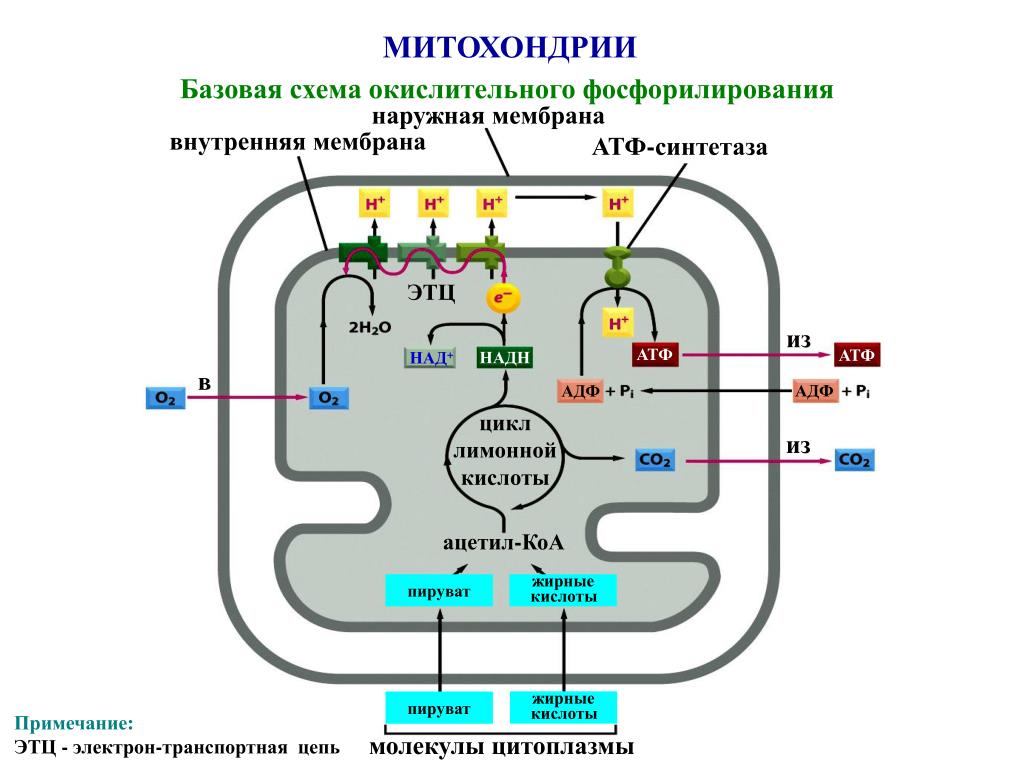
Сила, приводящая в движение «ротор» АТФ-синтазы, возникает при достижении разности потенциалов между наружной и внутренней сторонами мембраны > 220 мВ и обеспечивается потоком протонов, протекающих через специальный канал в F0, расположенный на границе между субъединицами a и c. При этом путь переноса протонов включает в себя следующие структурные элементы:
- Два расположенных несоосно «полуканала», первый из которых обеспечивает поступление протонов из межмембранного пространства к существенно важным функциональным группам F0, а другой обеспечивает их выход в матрикс митохондрии;
- Кольцо из субъединиц c, каждая из которых в своей центральной части содержит протонируемую карбоксильную группу, способную присоединять H+ из межмембранного пространства и отдавать их через соответствующие протонные каналы. В результате периодических смещений субъединиц с, обусловленных потоком протонов через протонный канал происходит поворот субъединицы γ, погружённой в кольцо из субъединиц с.

Таким образом, каталитическая активность АТФ-синтазы непосредственно связана с вращением её «ротора», при котором поворот субъединицы γ вызывает одновременное изменение конформации всех трёх каталитических субъединиц β, что в конечном счёте и обеспечивает работу фермента. При этом в случае образования АТФ «ротор» крутится по часовой стрелке со скоростью четыре оборота в секунду, а само подобное вращение происходит дискретными скачками по 120°, каждый из которых сопровождается образованием одной молекулы АТФ.
Непосредственная функция синтеза АТФ локализована на β-субъединицах сопрягающего комплекса F1. При этом самым первым актом в цепи событий, приводящих к образованию АТФ, является связывание АДФ и фосфата с активным центром свободной β-субъединицы, находящейся в состоянии 1. За счёт энергии внешнего источника (тока протонов) в комплексе F1 происходят конформационные изменения, в результате которых АДФ и фосфат становятся прочно связанными с каталитическим центром (состояние 2), где становится возможным образование ковалентной связи между ними, ведущей к образованию АТФ. На данной стадии АТФ-синтазы ферменту практически не требуется энергии, которая будет необходима на следующем этапе для освобождения прочно связанной молекулы АТФ из ферментативного центра. Поэтому следующий этап работы фермента заключается в том, чтобы в результате энергозависимого структурного изменения комплекса F1 каталитическая β-субъединица, содержащая прочно связанную молекулу АТФ, перешла в состояние 3, в котором связь АТФ с каталитическим центром ослаблена. В результате этого молекула АТФ покидает фермент, а β-субъединица возвращается в исходное состояние 1, благодаря чему обеспечивается цикличность работы фермента.
На данной стадии АТФ-синтазы ферменту практически не требуется энергии, которая будет необходима на следующем этапе для освобождения прочно связанной молекулы АТФ из ферментативного центра. Поэтому следующий этап работы фермента заключается в том, чтобы в результате энергозависимого структурного изменения комплекса F1 каталитическая β-субъединица, содержащая прочно связанную молекулу АТФ, перешла в состояние 3, в котором связь АТФ с каталитическим центром ослаблена. В результате этого молекула АТФ покидает фермент, а β-субъединица возвращается в исходное состояние 1, благодаря чему обеспечивается цикличность работы фермента.
Работа АТФ-синтазы связана с механическими движениями её отдельных частей, что позволило отнести этот процесс к особому типу явлений, названных «вращательным катализом». Подобно тому, как электрический ток в обмотке электродвигателя приводит в движение ротор относительно статора, направленный перенос протонов через АТФ-синтетазу вызывает вращение отдельных субъединиц фактора сопряжения F1 относительно других субъединиц ферментного комплекса, в результате чего это уникальное энергообразующее устройство совершает химическую работу — синтезирует молекулы АТФ. В дальнейшем АТФ поступает в цитоплазму клетки, где расходуется на самые разнообразные энергозависимые процессы. Подобный перенос осуществляется специальным встроенным в мембрану митохондрий ферментом АТФ/АДФ-транслоказой, который обменивает вновь синтезированную АТФ на цитоплазматическую АДФ, что гарантирует сохранность фонда адениловых нуклеотидов внутри митохондрий.
Митохондрии и наследственность
ДНК митохондрий наследуются почти исключительно по материнской линии. Каждая митохондрия имеет несколько участков нуклеотидов в ДНК, идентичных во всех митохондриях (то есть в клетке много копий митохондриальных ДНК), что очень важно для митохондрий, неспособных восстанавливать ДНК от повреждений (наблюдается высокая частота мутаций). Мутации в митохондриальной ДНК являются причиной целого ряда наследственных заболеваний человека.
См. также
Литература
- М. Б. Беркинблит, С. М. Глаголев, В. А. Фуралев. Общая биология. — М.: МИРОС, 1999.

- Д. Тейлор, Н. Грин, У. Стаут. Биология. — М.: МИР, 2006.
- Э. Уиллет. Генетика без тайн. — М.: ЭКСМО, 2008.
- Д. Г. Дерябин. Функциональная морфология клетки. — М.: КДУ, 2005.
Примечания
- ↑ [1] Энергетика клетки объяснила тайну появления сложных форм жизни, 25 октября 2010 membrana
Ссылки
(PDF) ПРАКТИЧЕСКАЯ МИТОХОНДРИОЛОГИЯ
103
проводимости внутренней мембраны митохондрий. Очевидно, ЭДТА удалял ионы Mg2+ с каких-
то мест на внутренней мембране с высоким сродством к магнию. В результате интегральный белок
менял свою конформацию и открывал пору для протонов и, возможно, для ионов K+. Пока не
известно, какой белок участвует в этом феномене. Вполне вероятно, что этот же белок может
участвовать в регуляции Са-зависимой поры проницаемости.
Первое сообщение о способности ЭДТА увеличивать дыхание в МС-4 было представлено в
2000 году Cadenas & Brand, 2000.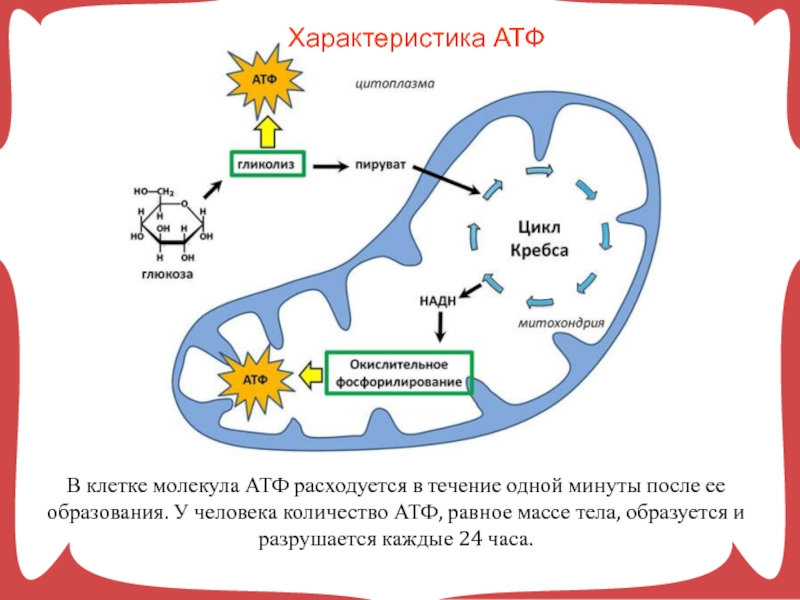 Эти авторы изучали эфекты ЭДТА и Mg2+ на потребление
Эти авторы изучали эфекты ЭДТА и Mg2+ на потребление
кислорода в МС-4, и пришли к выводу, что это Mg2+, а не пуриновые нуклеотиды (ГДФ, АТФ и
АДФ) контролируют протонную проводимость внутренней мембраны митохондрий (ВММ).
Cadenas & Brand (2000) не изучали изменения мембранного потенциала, и не использовали другие
субстраты, помимо сукцинат + ротенон. Нужно упомянуть, однако, что протонная поводимость
ВММ на самом деле контролируется также АДФ на уровне переносчика адениннуклеотидов
(Panov et al., 1980). Вероятно, что этот механизма в физиологических условиях играет более
значительную роль в регуляции мембранного потенциала через проводимость ионов Н+ и К+, чем
Mg2+, поскольку его концентрация сильно забуферена.
В литературе имеется много примеров необдуманного применения ЭДТА, что приводило к
неправильным результатам. Sorgato et al. (1974) были одними из первых, кто изучал продукцию
свободных радикалов кислорода (СРК) митохондриями мозга.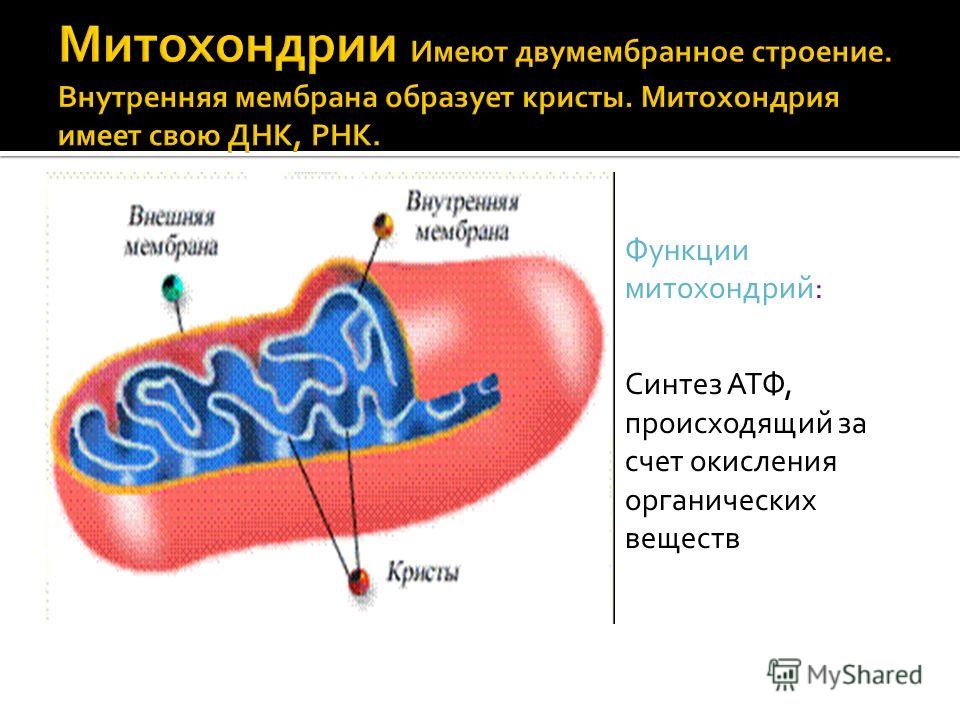 Эти авторы пришли к выводу, что в
Эти авторы пришли к выводу, что в
отличие от митохондрий сердца и печени, митохондрии мозга не продуцируют h3O2 или
супероксидный радикал (Sorgato et al., 1974). Этот ошибочный вывод на много лет замедлил
изучение продукции СРК митохондриями мозга, которые на самом деле являются одним из
важнейших патофизиологических механизмов развития нейродегенеративных заболеваний
(болезни Паркинсона, Алцгеймера, Амиотрофического бокового склероза и др.). Кто в «зравом
уме» стал бы тратить время на изучение того, о чем исследователи одной из наиболее уважаемых
лабораторий Мира, руководимой профессором Ацци (Azzi), показали, что оно не существует? Со
временем, однако, люди забыли об этой публикации, но скорее даже не чители ее. Опять же, кто
из молодых и «умных» будет читать «старые» публикации 10-20-летней давности? Однако, я
прочитал статью Sorgato et al. (1974), и поскольку у меня уже были на руках результаты
экспериментов, показанные на рисунке 7.![]() 2, то мне сразу стала понятна причина отрицательного
2, то мне сразу стала понятна причина отрицательного
вывода о способности митохондрий мозга продуцировать СРК. В экспериментах Sorgato et al.
(1974) среда выделения и инкубации митохондрий содержали 2 мМ ЭДТА, и Mg2+ не добавлялся
в среду, а в качестве субстрата использовали сукцинат. Как мы увидим позже, в митохондриях
мозга (и не только) основным источником СРК является обратный перенос электронов. Поскольку
в присутствии ЭДТА мембранный потенциал падал на 30 мВ, то при окислении сукцината не было
обратного переноса электронов и сопряженного сним образования СРК. Впрочем, в 1974 году еще
никто не знал о возможных последствиях применения ЭДТА, которые были описаны выше.
Поэтому, неправильный вывод Sorgato et al. (1974) вполне понятен и простителен. Ведь если ты
работаешь на границе неведомого, то неизбежно попадаешь в такие ситуации. А вот в наше время,
когда исследователи непродуманно используют ЭДТА и ЭГТА, такие ошибки уже
непростительны. А примеров довольно много. Но, пальцем указывать не буду.
А примеров довольно много. Но, пальцем указывать не буду.
Важно. Приведенный пример показывает, что использовать хелаторы двухвалентных
катионов нужно продуманно. Если по какой-то причине Вам нужно использовать 0.5-1.0 мМ
ЭДТА, например при загрязнении воды и химикатов ионами Cu2+, Zn2+ or Fe2+, то достаточно
добавить в среду выделения или инкубации 0.5 мМ MgCl2, и «разобщающий» эффект ЭДТА на
митохондрии будет устранен.
Скайраннинг — советы чайникам | АЛЬПИНДУСТРИЯ
17 ноября 2011 | Иван Григорьев
Советы и инструкции
Начало
Часть II
Продолжаю серию статей, посвящённых физиологии скайраннинга. Сегодня мы рассмотрим классификацию мышечных волокон и их изменение под воздействием различных видов нагрузки. К сожалению, многие люди не знакомы с этой информацией. Это очень грустно, потому что альпинисты должны быть сведущи в вопросах физиологии и подготовки.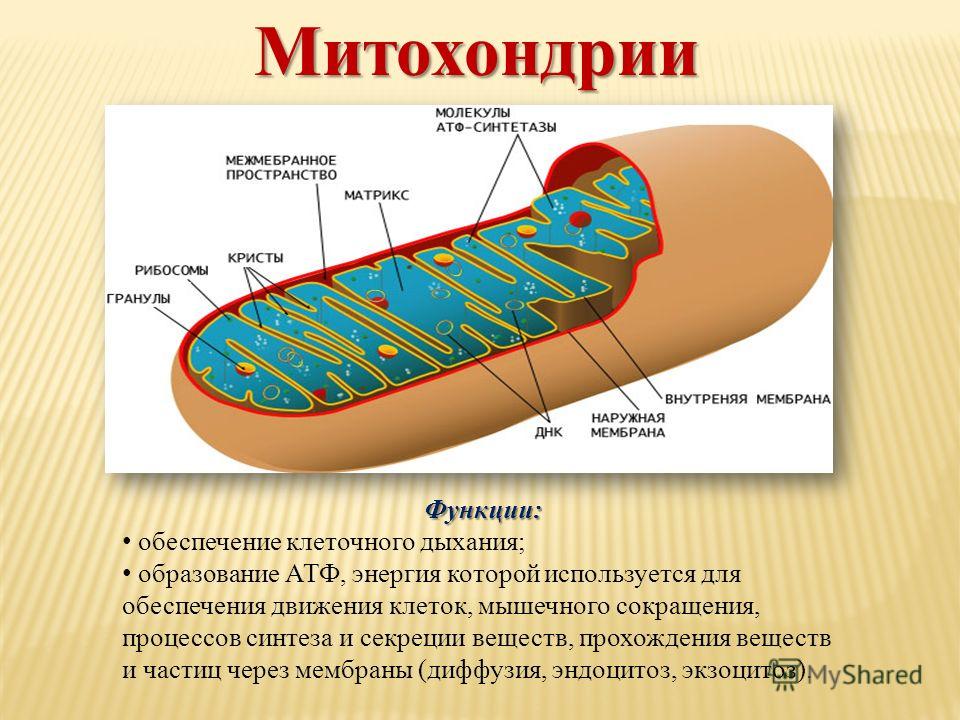
Что такое альпинизм – это в первую очередь выносливость, терпилово, акклиматизация, технические прибамбасы. Всё это невозможно без грамотной подготовки — скалолазной, велосипедной, беговой, лыжной, кому как приятнее, но бег — это всё-таки основа. С помощью бега вы всегда сможете подготовить своё сердце и мышцы к любому восхождению. Вы можете лазать 8а, это даст вам преимущество на скальном маршруте, но в горы всё-таки ходят. Именно поэтому я так надеюсь, что скайраннинг станет локомотивом профессиональной горной подготовки, так же, как в своё время стали скалолазание и ледолазание в подготовке к прохождению более сложных участков маршрутов.
Информация, публикуемая ниже, написана замечательным физиологом, нашим соотечественником Виктором Николаевичем Селуяновым. Его статьи доступны в интернете, как и большинство других материалов на тему физиологии и тренировок, но к сожалению, человек так устроен, что часто не в состоянии найти нужную информацию: некоторые материалы кажутся слишком сложными, некоторые слишком объёмными или простыми.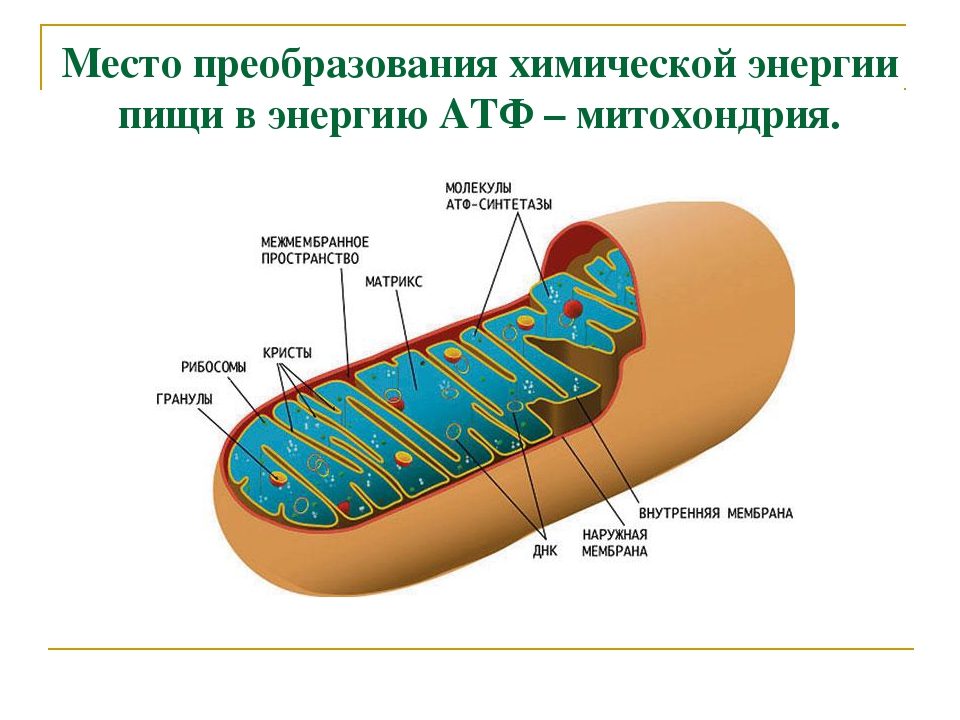 Так произошло с моим тренером, который 12 лет бегал марафоны, «убивал» себя и не имел ни малейшего представления о физиологии. Он знал что такое интервальная работа, пано, темповик, углеводный обмен, длительная, в каких объёмах и когда нужно выполнять, но всё это происходило на уровне подсознания. Несколько лет назад он нашёл Селуяновские статьи, точнее их порекомендовала Ирина Реутович, которая тренировалась – нет, не по ним, Ирина Владимировна тренировалась всегда по интуиции, у неё очень сильный организм. А статьи она дала для того, чтобы Михаил Викторович (мой тренер) «разобрался» в них. Вот так, всей компанией мы стали зубрить термины и купили пульсометры. Результат оказался потрясающим.
Так произошло с моим тренером, который 12 лет бегал марафоны, «убивал» себя и не имел ни малейшего представления о физиологии. Он знал что такое интервальная работа, пано, темповик, углеводный обмен, длительная, в каких объёмах и когда нужно выполнять, но всё это происходило на уровне подсознания. Несколько лет назад он нашёл Селуяновские статьи, точнее их порекомендовала Ирина Реутович, которая тренировалась – нет, не по ним, Ирина Владимировна тренировалась всегда по интуиции, у неё очень сильный организм. А статьи она дала для того, чтобы Михаил Викторович (мой тренер) «разобрался» в них. Вот так, всей компанией мы стали зубрить термины и купили пульсометры. Результат оказался потрясающим.
Если раньше наши тренировки носили угнетающий характер, мы молотили, что есть мочи, считали, что этого не достаточно, и молотили ещё. То теперь, помимо того, что мы не чувствовали такой усталости, за счёт того, что грамотно распределяли нагрузку, наши результаты полезли вверх. Через несколько месяцев после начала тренировок по Селуянову мы уже могли бежать по 4 минуты на км на пульсе всего-лишь 140 ударов. Правда, потом мы стали готовиться к суточному бегу и в результате измотали себя, потому что не следили за мышцами, но уже тогда мы поняли что к чему. Именно поэтому я публикую то, что проверено мной и моими друзьями, на себе, за годы тренировок.
Правда, потом мы стали готовиться к суточному бегу и в результате измотали себя, потому что не следили за мышцами, но уже тогда мы поняли что к чему. Именно поэтому я публикую то, что проверено мной и моими друзьями, на себе, за годы тренировок.
Помимо Селуяновских статей я хотел порекомендовать ещё две книги: первая Питт Фитзингер и Скотт Дуглас «Бег по шоссе для серьёзных бегунов» и вторая Дэнни Дрэйер «Ци бег». Последнюю кстати я прочитал по рекомендации известного российского марафонца Леонида Швецова. В его словах звучали сожаления, что эта книга не попалась ему тогда, когда он только начинал свою беговую карьеру. Если у кого-то возникнут проблемы с поиском этих материалов, я могу выслать их по электронной почте, пишите.
А теперь, собственно, по делу.
Словарь чайника
МВ – мышечное волокно (волокна)
ММВ – медленные мышечные волокна
БМВ – быстрые мышечные волокна
ОМВ – окислительные мышечные волокна
ГМВ – гликолитические мышечные волокна
АэП – аэробный порог
АнП – анаэробный порог
МПК – максимальное потребление кислорода
КФ — креатинфосфат
АТФ – аденозинтрифосфорная кислота (основная «энергетическая валюта» клетки)
Миофибриллы — сократимые элементы мышечной клетки (цилиндрические нити толщиной 1 — 2 мкм, идущие вдоль от одного конца мышечного волокна до другого), сокращаются в присутствии АТФ.
Митохондрии – клеточные органеллы (элементы), в которых синтезируется АТФ за счет окислительного фосфорилирования.
Окислительное фосфорилирование – функция клеточного дыхания, при которой происходит синтез АТФ (идет в митохондриях).
Цикл Кребса (цикл трикарбоновых кислот, цикл лимонной кислоты) представляет собой серию химических реакций, протекающих в митохондриях, и является общим конечным путем окисления углеводов, липидов и белков.
Миокард – сердечная мышца
Миокардиоцит – клетка миокарда
Классификация мышечных волокон. Изменение мышечной композиции под действием тренировки
Остановимся подробнее на классификации мышечных волокон. Первый способ — на быстрые мышечные волокна (БМВ) и медленные мышечные волокна (ММВ), эта классификация идет по ферменту АТФаза миофибрилл (сократительных элементов), тип которого может быть быстрым или медленным. Отсюда быстро сокращающиеся и медленно сокращающиеся МВ.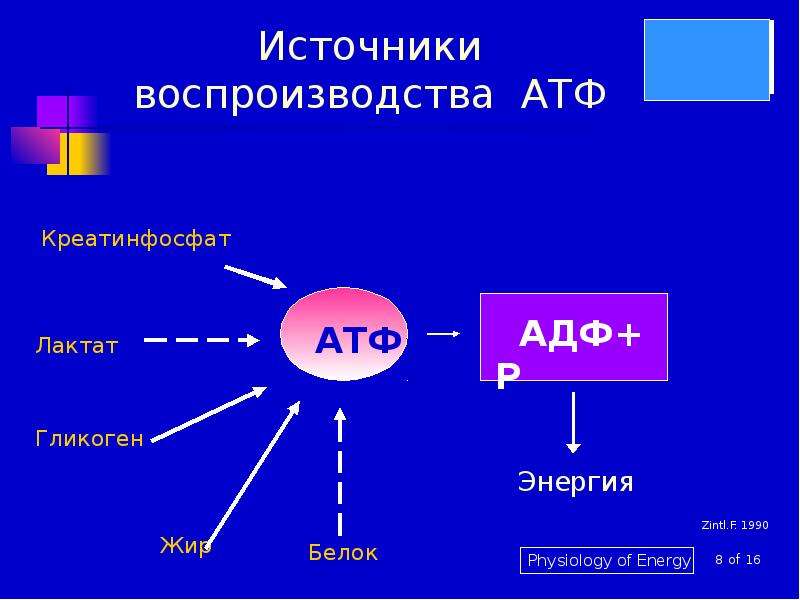 Соотношение быстрых и медленных волокон определяется наследственной информацией, и изменить его мы практически не можем.
Соотношение быстрых и медленных волокон определяется наследственной информацией, и изменить его мы практически не можем.
Второй способ – разделение МВ на окислительные и гликолитические, а они делятся уже не по миофибрилле, а по количеству митохондрий (структур клетки, где происходит потребление кислорода). Если есть митохондрии, то МВ окислительные, мало митохондрий или почти нет — гликолитические. Способность МВ к гликолизу также наследуется и определяется количеством ферментов гликолитического типа. Но вот количество митохондрий достаточно легко изменяется под воздействием тренировок. И с увеличением числа митохондрий МВ, бывшее гликолитическим, становится окислительным.
К сожалению, в этом вопросе существует путаница. Обычно смешивают обе классификации. Говорят о медленных, а подразумевают окислительные, смешивают гликолитические и быстрые. На самом деле медленные тоже могут быть гликолитическими, хотя этот вариант в литературе не описывается. Но мы знаем, что если человек лежит в больнице предоперационный период, а потом ещё и послеоперационный период, то потом уже и встать не может, ходить не может.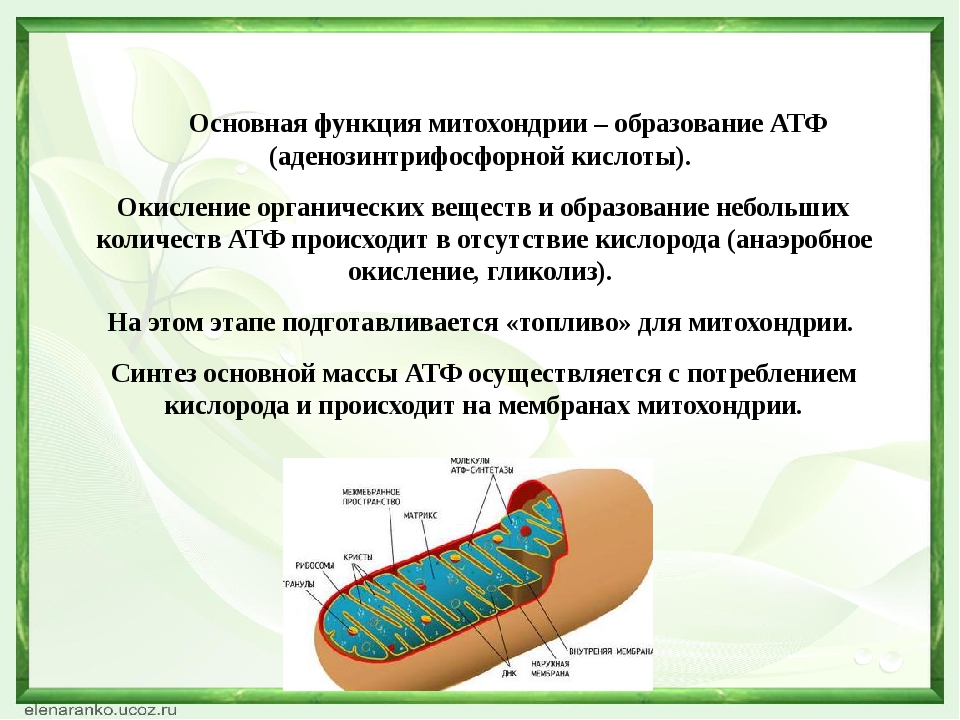 Первая причина — координация нарушается, а вторая причина — мышцы уходят. И самое главное, уходят, прежде всего, митохондрии из ММВ (период их «полураспада» всего 20 — 24 дня). Если человек пролежал 50 дней, то от митохондрий почти ничего не останется, МВ превратятся в медленные гликолитические, поскольку медленные или быстрые наследуется, а митохондрии создаются. (Быстрые МВ при правильных тренировках также могут стать окислительными).
Первая причина — координация нарушается, а вторая причина — мышцы уходят. И самое главное, уходят, прежде всего, митохондрии из ММВ (период их «полураспада» всего 20 — 24 дня). Если человек пролежал 50 дней, то от митохондрий почти ничего не останется, МВ превратятся в медленные гликолитические, поскольку медленные или быстрые наследуется, а митохондрии создаются. (Быстрые МВ при правильных тренировках также могут стать окислительными).
Поэтому с точки зрения тренировочного процесса для данного спортсмена не интересно деление МВ на медленные и быстрые – это имеет значение на этапе отбора. Вся логика построения тренировки идет не с точки зрения сокращения мышц по скорости, а направлена на превращение ГМВ в окислительные. Ибо в этом случае мы изменяем конкретного человека.
Цель тренировки в циклических видах спорта — создавать митохондрии. Только митохондрии потребляют кислород, значит, спортивная форма растет по мере накопления митохондрий. Возьмем мышечное волокно. У него есть миофибриллы, каждая миофибрилла оплетается митохондриями, и больше определенного предела они не могут образоваться, только в один слой, если условно так говорить.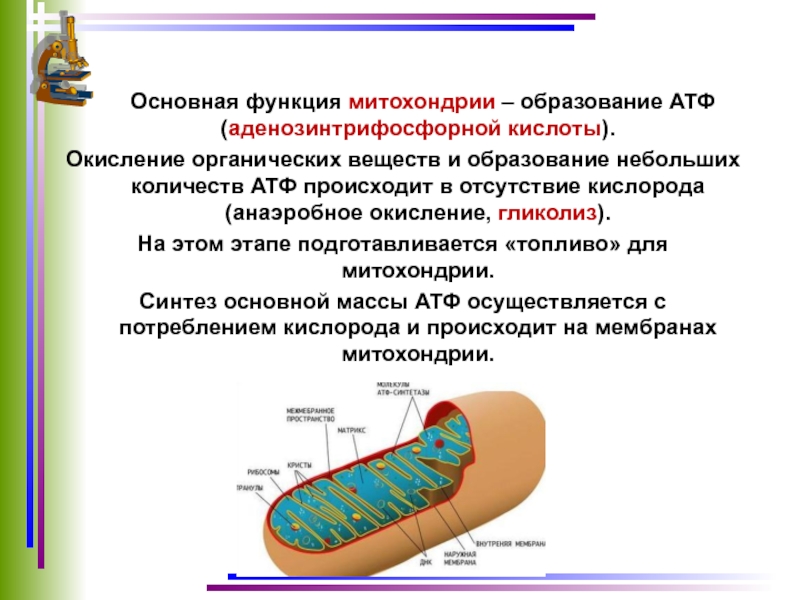 В конце концов, эти МВ накапливают столько митохондрий, что больше ничего прибавить не могут. ММВ быстро выходят на предел подготовленности, и дальше весь процесс роста спортивной формы идет через то, что мы гликолитические превращаем в окислительные. (Низкопороговые МВ потому и окислительные, что постоянно работают при любых интенсивностях с максимальной для них мощностью). Перестаём тренироваться или, например, начинаем низкопороговые тренировать, тогда высокопороговые митохондрии теряют. Весь смысл набора спортивной формы — набрать митохондрии в МВ высокопороговых двигательных единиц, другого пути нет. Все только этим и занимаются, а думают об интервальной тренировке и еще о чем-то, то есть о формальности. А суть тренировки — поменять содержание мышечных волокон, то есть добавить митохондрий.
В конце концов, эти МВ накапливают столько митохондрий, что больше ничего прибавить не могут. ММВ быстро выходят на предел подготовленности, и дальше весь процесс роста спортивной формы идет через то, что мы гликолитические превращаем в окислительные. (Низкопороговые МВ потому и окислительные, что постоянно работают при любых интенсивностях с максимальной для них мощностью). Перестаём тренироваться или, например, начинаем низкопороговые тренировать, тогда высокопороговые митохондрии теряют. Весь смысл набора спортивной формы — набрать митохондрии в МВ высокопороговых двигательных единиц, другого пути нет. Все только этим и занимаются, а думают об интервальной тренировке и еще о чем-то, то есть о формальности. А суть тренировки — поменять содержание мышечных волокон, то есть добавить митохондрий.
Вот вы начинаете правильно тренироваться и набираете митохондрий всё больше, больше и больше, мышцы переходят из формы гликолитической в окислительную, то есть с обилием митохондрий. И когда все мышечные волокна становятся окислительными — это предел спортивной формы, больше ничего не получится. Хотя тут есть одна хитрость. Дело в том, что окислительные волокна потребляют только жиры (пока есть запас жиров), а мощность при окислении жиров теряется. Отсюда получается некий парадокс — не надо делать так, чтобы мышцы были только окислительные, надо оставить немного гликолитических, иначе будете на жирах бежать, а мощность функционирования на жирах меньше примерно на 15%. Тогда те же самые мышцы будут более мощно работать. Понятно, что к лыжному спорту это тоже относится.
Хотя тут есть одна хитрость. Дело в том, что окислительные волокна потребляют только жиры (пока есть запас жиров), а мощность при окислении жиров теряется. Отсюда получается некий парадокс — не надо делать так, чтобы мышцы были только окислительные, надо оставить немного гликолитических, иначе будете на жирах бежать, а мощность функционирования на жирах меньше примерно на 15%. Тогда те же самые мышцы будут более мощно работать. Понятно, что к лыжному спорту это тоже относится.
Влияние гликолитических и окислительных мышечных волокон на результат
Так вот, вы начинаете бежать среднюю дистанцию, разбегаетесь, и выходите на порог анаэробного обмена, он как раз соответствует моменту, когда функционирует все ОМВ и даже часть гликолитических. При этом получается, что человек выходит на крейсерскую скорость. Если у него только ОМВ, то он так и будет стабильно молотить. Прибавить не может и убавить не может (убавить, конечно, может, но это ему не надо, а прибавить не может, потому что не чем добавить), он прибежит с той же самой скоростью на финиш. Если с ним будет бежать точно такой же человек, но у которого будет запас ГМВ, то он на финише всегда прибавит. Значит, получается, средневик — это человек, у которого есть запас мышечных волокон, которые он может включить в работу, и лучше быстрых гликолитических, тогда финиш будет еще быстрее. Так же и у лыжников: тот, у кого есть запас ГМВ, на финише выиграет, если дистанция будет ровная. Но, увы, так не бывает.
Если с ним будет бежать точно такой же человек, но у которого будет запас ГМВ, то он на финише всегда прибавит. Значит, получается, средневик — это человек, у которого есть запас мышечных волокон, которые он может включить в работу, и лучше быстрых гликолитических, тогда финиш будет еще быстрее. Так же и у лыжников: тот, у кого есть запас ГМВ, на финише выиграет, если дистанция будет ровная. Но, увы, так не бывает.
Снова перейдем на более простой вид спорта, велосипедный (мне ближе). Рассмотрим спортсмена, у которого ОМВ только 15-20%, остальные — гликолитические. На равнине он набирает критическую скорость, превышает её, и начинает постепенно закисляться. Проходит 5-6 минут, он попадает в мертвую точку, пульс запредельный, дышать невозможно. Спортсмен начинает мощность снижать, и через 2-3 км выходит, наконец, на ту самую скорость, которая нужна. Вот классический вариант развития физиологических процессов на равнине. А если это не равнина, а холмистая местность, и холмы короткие, по длине такие, что на подъем затрачивается не больше 30 секунд? Тогда в этот холм спортсмен включает свои ГМВ, их хватает ровно на 30 секунд.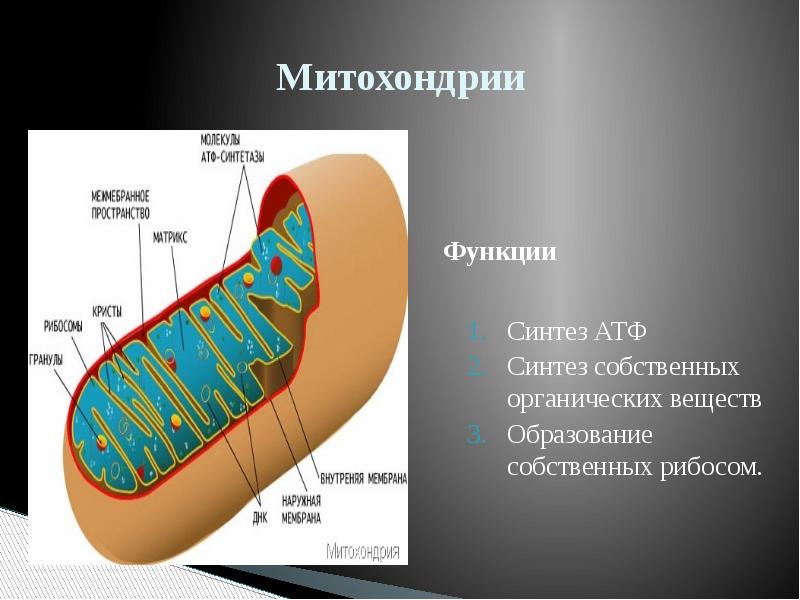 В холм влетает, скорость большая, а со спуска работать уже практически не надо, ГМВ восстанавливаются, потом опять подъём, спуск и т.д. При этом он может влететь в этот подъём быстро и мощно, а другой, у кого только одни окислительные, такой мощности не получит, попытается отыграть на спуске, но это очень трудно и особенно добавить не удастся. В этих условиях спортсмен, у которого много ГМВ, начинает выигрывать.
В холм влетает, скорость большая, а со спуска работать уже практически не надо, ГМВ восстанавливаются, потом опять подъём, спуск и т.д. При этом он может влететь в этот подъём быстро и мощно, а другой, у кого только одни окислительные, такой мощности не получит, попытается отыграть на спуске, но это очень трудно и особенно добавить не удастся. В этих условиях спортсмен, у которого много ГМВ, начинает выигрывать.
Рассмотрим двух спортсменов в равных условиях, но у первого мышцы покрупнее (больше ГМВ), а у второго поменьше. Если это равнина, первый, скорее всего, выиграет, потому что включит на финише гликолитические волокна. По дистанции они будут одинаково ехать, а на финише первый выиграет с разницей в 1-2 секунды. Если холмистая местность, но с короткими холмами, выиграет первый, у которого больше гликолитических МВ, может еще больше выиграет, потому что он в каждую горку 1-2 секунды отыграет, а со спуска еще быстрее уедет. Но как только горка превращается в минутную, то на первой он 2 секунды отыграет за 30 секунд, второй немножко отстал, а потом на следующей горке второй ему 10 секунд ввезет, потому что у первого ГМВ перестанут нормально работать, закислятся, а у второго ничего не закисляется, он со стабильной скоростью до верха и доедет.
Вот тут эти нюансы и возникают.
Теперь переключимся на лыжи. Если спринт будет с короткими подъёмами или же длинная дистанция с короткими подъёмами, выиграет тот, у кого есть запас ГМВ и очень большой. Но в лыжном спорте коротких подъемов почти не бывает. А как только подъёмы по длительности уходят за 30 секунд, всё меняется, к 40-й секунде ноги начинают здорово болеть, а к 1 минуте дыхание резко учащается, потому что ГМВ начинают накапливать ионы водорода, молочную кислоту, начинается значительное выделение углекислого газа, он заставляет интенсивно дышать, пульс за 200 и страшные мучения. Если всё время выходить на пульс 200-240, повторять его по ходу гонки 10 – 15 — 20 раз, то и соперника не увидишь… (состояние будет предельно тяжелым).
Физиология мышечного сокращения. Закон рекрутирования мышечных волокон
Напомним современные знания физиологии мышечного сокращения. Начнем с учебных знаний. В учебнике пишется, что существует процесс сокращения мышцы, и он обеспечивается некими механизмами энергообеспечения. Сам механизм сокращения связан с затратой молекул АТФ, молекулы АТФ должны быть внутри синтезированы с помощью молекулы КФ, а свободный креатин и свободный фосфат являются стимулом для разворачивания либо анаэробного гликолиза, либо аэробного гликолиза, либо окисления жиров. Вот классическая схема, современная, которая сейчас принята. Эта уточненная схема предложена Саксом, нашим ученым (у Чазова работает), для миокарда. В схеме существует КФ шунт, или, другими словами, все метаболические и энергетические пути, гликолиз и окисление жиров идут только через ресинтез КФ, а уже КФ идет непосредственно на ресинтез АТФ. Вот современные учебные знания.
Сам механизм сокращения связан с затратой молекул АТФ, молекулы АТФ должны быть внутри синтезированы с помощью молекулы КФ, а свободный креатин и свободный фосфат являются стимулом для разворачивания либо анаэробного гликолиза, либо аэробного гликолиза, либо окисления жиров. Вот классическая схема, современная, которая сейчас принята. Эта уточненная схема предложена Саксом, нашим ученым (у Чазова работает), для миокарда. В схеме существует КФ шунт, или, другими словами, все метаболические и энергетические пути, гликолиз и окисление жиров идут только через ресинтез КФ, а уже КФ идет непосредственно на ресинтез АТФ. Вот современные учебные знания.
В соответствии с ними, если спортсмен начинает двигаться в режиме «во всю», в течение примерно 15 секунд тратятся запасы АТФ и КФ (фосфагенов). Потом должен развернуться процесс, который стимулируется свободным креатином. Это, в первую очередь, процесс анаэробного гликолиза, который продолжается одну, может быть, полторы минуты, а вслед за этим должен развернуться процесс окислительного фосфорилирования, то есть начинается уже аэробный гликолиз.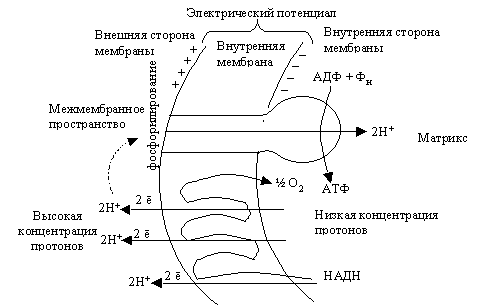 У нормального человека запасы углеводов снижаются где-то после 20-30 мин, а полностью заканчиваются через 45 мин. И только тогда, когда заканчиваются запасы углеводов в мышце и глюкоза в крови, начинает интенсивно развиваться процесс, связанный с окислением жиров. В случае передвижения со средней интенсивностью, при недостатке кислорода в крови, разворачивается анаэробный гликолиз. Это классическая схема.
У нормального человека запасы углеводов снижаются где-то после 20-30 мин, а полностью заканчиваются через 45 мин. И только тогда, когда заканчиваются запасы углеводов в мышце и глюкоза в крови, начинает интенсивно развиваться процесс, связанный с окислением жиров. В случае передвижения со средней интенсивностью, при недостатке кислорода в крови, разворачивается анаэробный гликолиз. Это классическая схема.
Но эта схема не выдерживает критики, когда мы переходим с представлений уровня пробирки или одного единственного мышечного волокна к мышце в целом. Для единственного изолированного МВ это более или менее правильное описание. Но у нас не одно МВ, а множество, еще есть множество мышц и, следовательно, в нашу модель мы должны включить и эти элементы. Кроме того, у нас есть ОМВ и ГМВ, у нас есть те МВ, которые раньше рекрутируются при определенной интенсивности: если интенсивность меняется, то дополнительные мышечные волокна включаются. Короче говоря, есть закон рекрутирования МВ.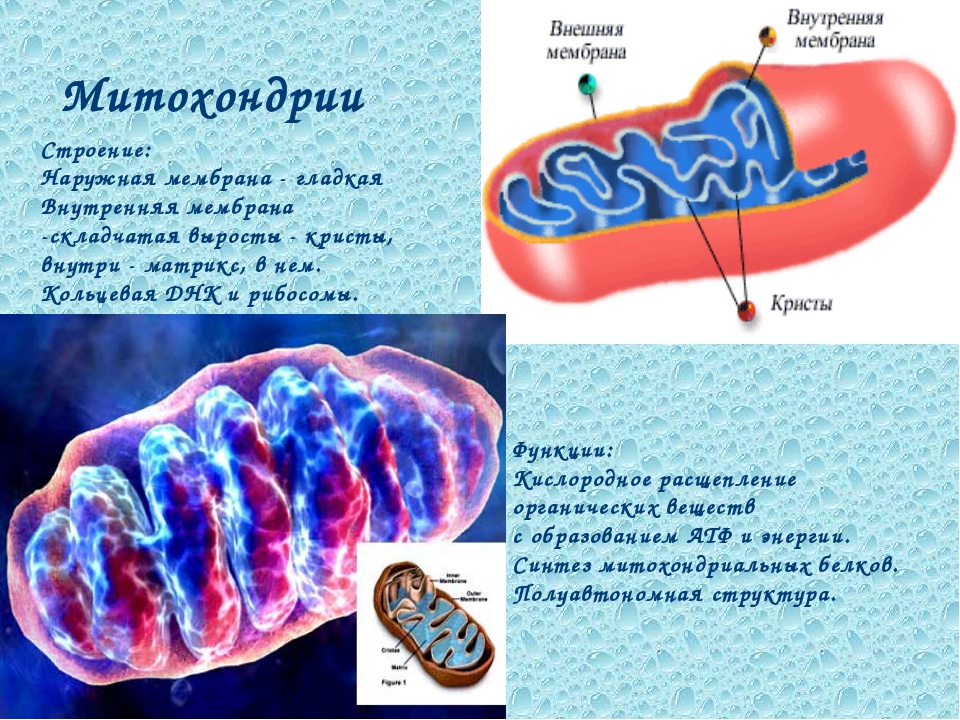 Если все эти компоненты учесть, то мы построим новую модель, которая состоит из центральной нервной системы, которая управляет мотонейронами в спинном мозге, а мотонейроны управляют мышцами. И вот в зависимости от импульсации, которая идет сверху, рекрутируются сначала низкопороговые двигательные единицы, а потом всё более высокопороговые, когда, допустим, увеличивается сила отталкивания. И в этом случае получается совсем другая картина.
Если все эти компоненты учесть, то мы построим новую модель, которая состоит из центральной нервной системы, которая управляет мотонейронами в спинном мозге, а мотонейроны управляют мышцами. И вот в зависимости от импульсации, которая идет сверху, рекрутируются сначала низкопороговые двигательные единицы, а потом всё более высокопороговые, когда, допустим, увеличивается сила отталкивания. И в этом случае получается совсем другая картина.
Например, вы начинаете двигаться с усилием 50% от максимума, максимум — это спринт (3-7 секунд), а 50% — это, условно говоря, бег на 1500 м или на 3000 м. Что будет происходить в организме? Вы рекрутируете столько мышечных волокон, сколько необходимо, чтобы держать скорость. Допустим, у вас 75% ОМВ. Допустим, вы рекрутировали половину всех мышечных волокон. Рекрутированные ОМВ отрабатывают 15 сек за счет АТФ и КФ, затем мощность их начинает падать где-то наполовину, и дальше эти ОМВ работают только в аэробном режиме, и пока используют только жиры.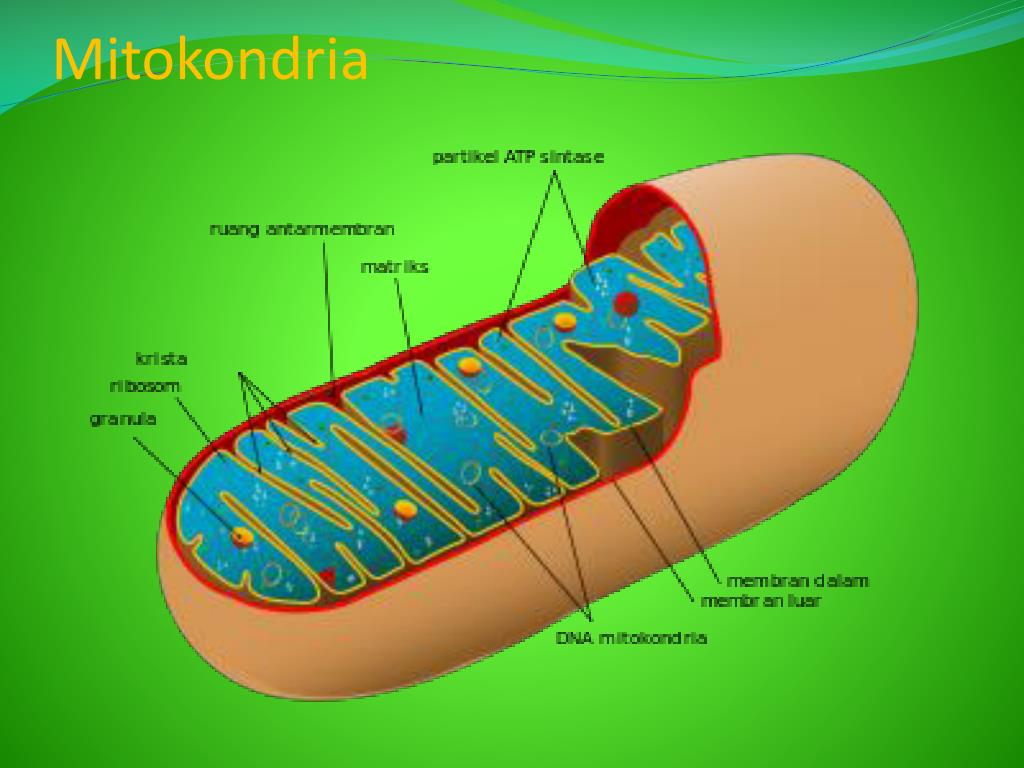 Не через 40 минут, а прямо сейчас, на 1-й минуте будут работать за счет окисления жиров! Потому что в ОМВ митохондрии, когда работают, выделяют наружу цитрат, который ингибирует (подавляет) гликолиз, поэтому могут окисляться только жиры (химию процесса окисления описывает цикл Кребса). Значит, не прошло и 15 секунд, как начали окисляться жиры. И вот мощность упала, а вам-то задание держать 50% от максимальной. Тогда вы обязаны рекрутировать еще порцию мышечных волокон. Допустим, дополнительные 25% вы рекрутируете, тоже окислительные, только они еще не работали, и они свои первые 15 секунд отрабатывают на АТФ и КФ. Получается, что на АТФ и КФ вы бежите уже не 15 секунд, а 30. То есть вы 15 секунд бежали на АТФ рекрутированных вначале МВ, и еще 15 секунд на следующих, но часть работы уже выполняется за счет аэробной продукции. Эти окислительные включились в работу, истратили свои запасы АТФ и КФ, не полностью, а наполовину, а вот эта половина поддерживается за счёт ресинтеза, то есть уже за счёт окислительных процессов, за счет жиров.
Не через 40 минут, а прямо сейчас, на 1-й минуте будут работать за счет окисления жиров! Потому что в ОМВ митохондрии, когда работают, выделяют наружу цитрат, который ингибирует (подавляет) гликолиз, поэтому могут окисляться только жиры (химию процесса окисления описывает цикл Кребса). Значит, не прошло и 15 секунд, как начали окисляться жиры. И вот мощность упала, а вам-то задание держать 50% от максимальной. Тогда вы обязаны рекрутировать еще порцию мышечных волокон. Допустим, дополнительные 25% вы рекрутируете, тоже окислительные, только они еще не работали, и они свои первые 15 секунд отрабатывают на АТФ и КФ. Получается, что на АТФ и КФ вы бежите уже не 15 секунд, а 30. То есть вы 15 секунд бежали на АТФ рекрутированных вначале МВ, и еще 15 секунд на следующих, но часть работы уже выполняется за счет аэробной продукции. Эти окислительные включились в работу, истратили свои запасы АТФ и КФ, не полностью, а наполовину, а вот эта половина поддерживается за счёт ресинтеза, то есть уже за счёт окислительных процессов, за счет жиров. И при заданной 50-процентной мощности вы обеспечиваете где-то 30-35% за счет окислительного фосфорилирования. При такой мощности где-то через 30-40 секунд вы выходите на предельные возможности этой мышцы в потреблении кислорода (она равна как раз 35% от максимальной мощности, которую эта мышца может развить). Это соответствует как раз АнП. Если нарисовать кривую потребления кислорода, то вы обнаружите плато, которое будет соответствовать АнП уже через 40 сек.
И при заданной 50-процентной мощности вы обеспечиваете где-то 30-35% за счет окислительного фосфорилирования. При такой мощности где-то через 30-40 секунд вы выходите на предельные возможности этой мышцы в потреблении кислорода (она равна как раз 35% от максимальной мощности, которую эта мышца может развить). Это соответствует как раз АнП. Если нарисовать кривую потребления кислорода, то вы обнаружите плато, которое будет соответствовать АнП уже через 40 сек.
Далее спортсмен будет рекрутировать ГМВ, но маленькими порциями, исходя из нормы мощности, которую вы задали. Вот он в течение минуты будет рекрутировать гликолитические. Они тоже сначала на АТФ и КФ работают, а потом за счет гликолиза. Включенные ГМВ минуту отработают, закислятся и снизят мощность почти до нуля. Поэтому придется включать новые ГМВ до тех пор, пока у вас есть их запас. Если он у вас большой, то можно минуты 3-4 так поработать. А тот, у кого запаса ГМВ нет, начнет снижать мощность, и откажется от выполнения задания.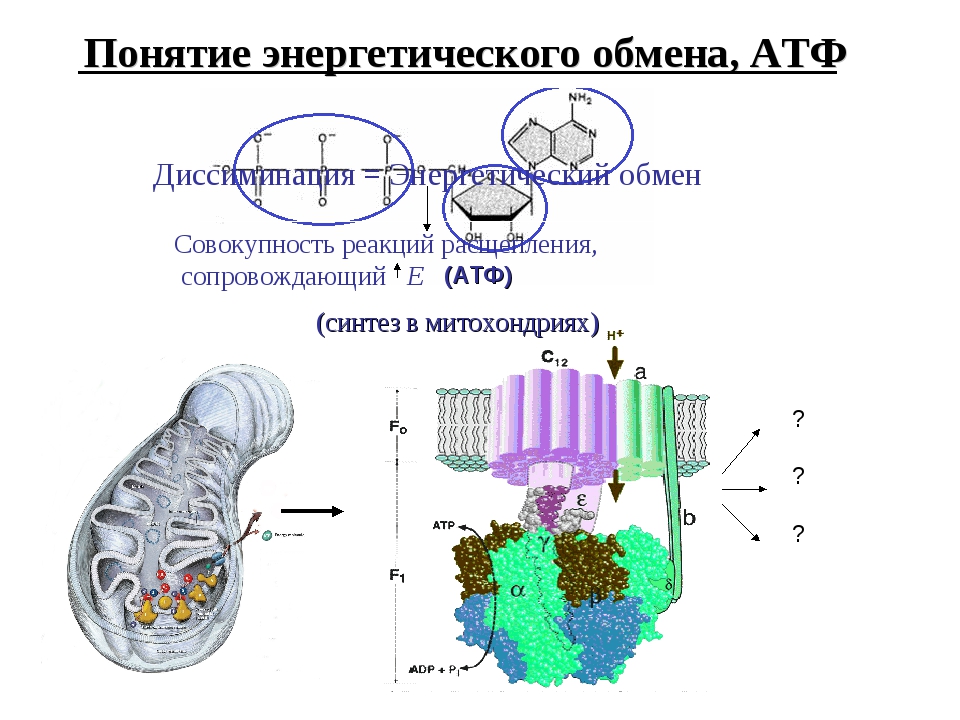
В итоге у тех, у кого ОМВ много, а гликолитических мало, кривая мощности поднимется, продержится где-то минуты полторы – две, и обязательно упадет на уровень АнП, и так будет держаться долго. Тот, кто имеет больший запас ГМВ, при прочих равных условиях сможет дольше проработать на высокой мощности, и на какой-то определенной дистанции выиграет. Получается, что человек, имеющий много ГМВ, но мало окислительных, на относительно коротких дистанциях, допустим, 1 — 1,5 минуты, ещё может выигрывать запасом гликолитических. Но чем длиннее дистанция, тем менее важна вот эта лишняя мышечная масса (ГМВ). И когда время на дистанции уходит, допустим, за 5 минут, то получается, что надо на себе везти лишнюю массу.
Из-за чего появляется специализация
В велосипедном спорте на равнине лишний вес не имеет принципиального значения. А если это гора, то даже в велосипедном спорте начинает играть роль собственный вес, спортсмен начинает тратить энергию на то, чтобы везти в подъем лишнюю мышечную массу.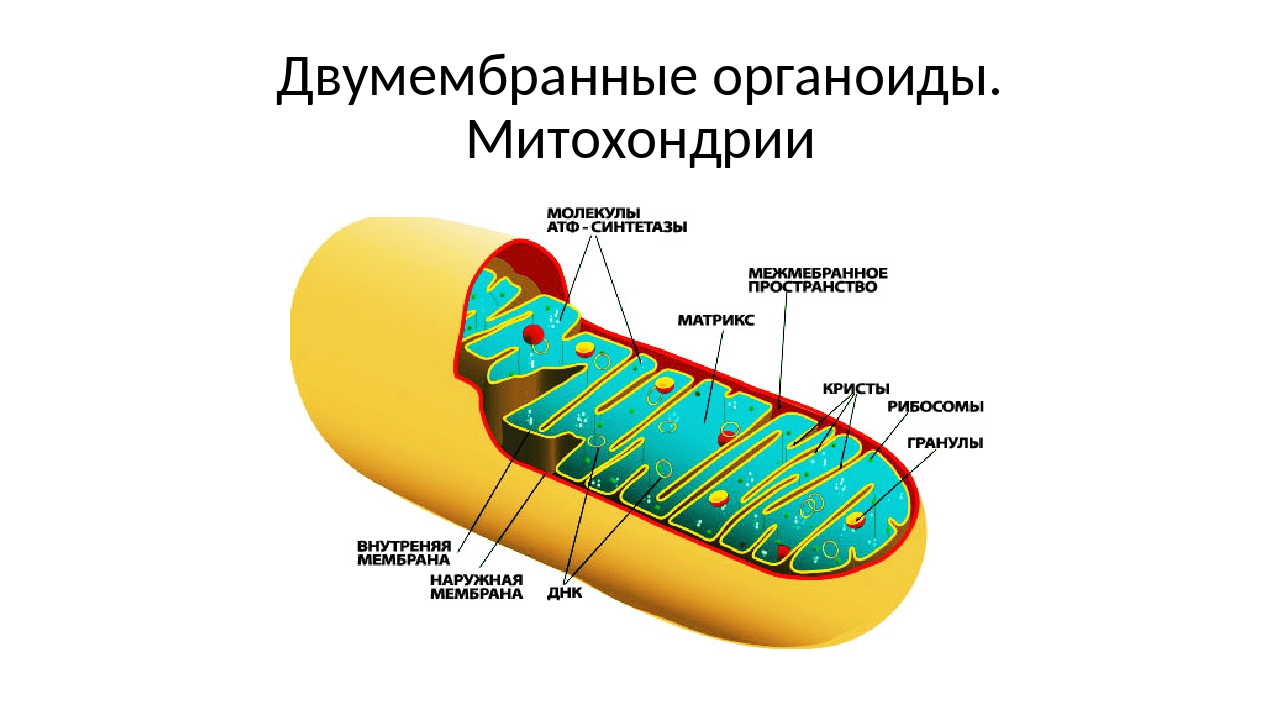 Поэтому чем длиннее дистанция, тем «вреднее» эта лишняя масса, и надо от нее всеми способами избавляться.
Поэтому чем длиннее дистанция, тем «вреднее» эта лишняя масса, и надо от нее всеми способами избавляться.
То же и в конькобежном спорте. Конечно, спортсмен в основном работает против ветра, но еще надо много энергии тратить, чтобы перемещать свое тело поперек дорожки, держать позу, а именно — везти свой собственный вес. Значит, и здесь вес начинает играть свою роль. Поэтому, если конькобежец везет на себе лишнюю мышечную массу, она мешает. На дистанциях в 500 и 1000 м некоторая «лишняя» масса помогает, потому что мощный толчок помогают сделать еще и мышцы рук и туловища. Но чем длиннее дистанция, тем больше «лишняя» масса мешает. Поэтому там, где возникают проблемы «лишней» массы, и появляется какая-то специализация (спринтер — стайер). Но иногда это не принципиально, если у спортсмена сильные мышцы ног с большой долей ОМВ (как у Хайдена).
Как и везде, существует простая модель и сложная. В сложной модели, вы видите, процессы по-другому разворачиваются, и даже можно объяснить, зачем нужны гликолитические волокна.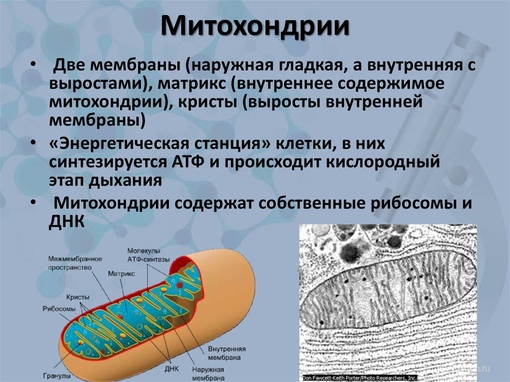 Пока дистанция относительно короткая, и если эта лишняя масса не очень мешает, то это очень выгодно. Чем длиннее дистанция и чем больше нагрузка, связанная с преодолением собственного веса, тем вреднее становится избыток ГМВ.
Пока дистанция относительно короткая, и если эта лишняя масса не очень мешает, то это очень выгодно. Чем длиннее дистанция и чем больше нагрузка, связанная с преодолением собственного веса, тем вреднее становится избыток ГМВ.
Центральные и периферические аэробные компоненты, их вклад в работоспособность
Теперь рассмотрим зависимость работоспособности от центрального и периферического факторов (сердечно-сосудистой системы и мышц). Если рассматривать какое-то конкретное двигательное действие — велосипед, коньки, легкую атлетику (бег) или лыжи, то мы увидим, что в каждом конкретном упражнении участвуют определенные мышечные группы. Если посчитать их массу, то окажется, что в велосипедном спорте одна мышечная масса, в легкой атлетике побольше, а в лыжном спорте еще больше. Возникает вопрос: сколько эти мышцы потребляют кислорода? Чисто теоретически это очень просто посчитать: 1 кг мышечной массы, если она находится на пределе подготовленности, потребляет кислорода где-то 0,2-0,3 л/мин, если в работе участвуют все ОМВ. Дальше надо просто умножить эту цифру на ту массу, которая есть, при условии, что она максимально подготовлена. Что значит максимально подготовлена?
Дальше надо просто умножить эту цифру на ту массу, которая есть, при условии, что она максимально подготовлена. Что значит максимально подготовлена?
Внутри этой мышечной массы одни ОМВ, миофибриллы и митохондрии находятся в таком соотношении, что больше уже ничего прибавить нельзя (миофибриллы все оплетены митохондриями, как в миокарде). И тогда получается, что для потребления 3 л кислорода нужно иметь 10 кг активной мышечной массы, а если нужно потреблять 6 л — достаточно иметь только 20 кг активной мышечной массы.
Теперь посчитаем, сколько кислорода может доставить сердце. Если принять, что 1 литр крови переносит 160 мл кислорода (при нормальном уровне гемоглобина), то, умножив это количество на минутный объём кровообращения, мы получим возможности сердца по доставке кислорода. У обычного человека, мужчины, ударный объём составляет порядка 120-130 мл за один выброс крови. При пульсе 190 ударов в минуту получим 190 уд/мин * 130 мл * 160 мл = около 4 л/мин. Всё так и считается, достаточно просто.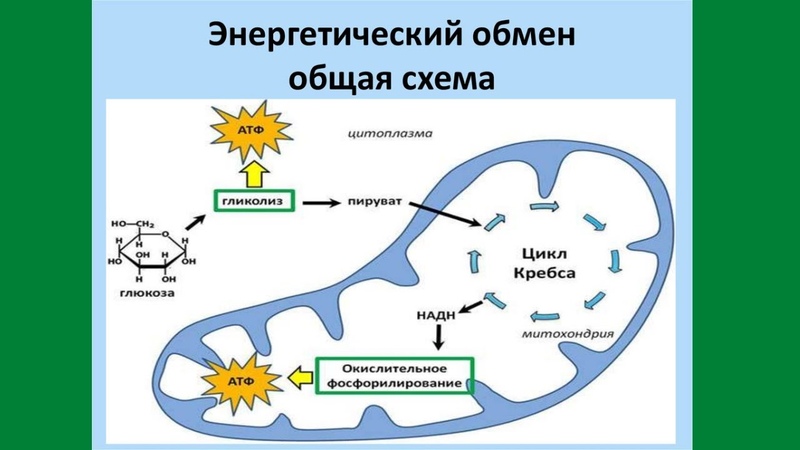 У супер-атлетов за один ударный цикл выбрасывается 240 мл, это соответствует 7-8 л/мин кислорода.
У супер-атлетов за один ударный цикл выбрасывается 240 мл, это соответствует 7-8 л/мин кислорода.
Мы определили, что 20 кг мышечной массы могут потребить около 6 литров кислорода в минуту. Если у лыжника на ногах мышечная масса 20 — 25 кг, и к этому добавить мышцы живота, спины, рук, то мы уйдем за цифру 30 с лишним килограммов. Сделаем поправку на то, что не вся эта мышечная масса будет потреблять кислород на пределе возможностей, и получим, что 40 кг активных мышц могут потребить кислорода около 8 л/мин. Вот столько должно перекачать сердце, чтобы полностью обеспечить мышцы кислородом, если эти мышцы максимально готовы.
Таким образом, мы получили два предела. Первый — из литературы известно, что перекачать 8 л/мин кислорода через организм с помощью сердца — это цифра предельная, этой цифры практически ни у кого нет. В то же время, 8 л/мин кислорода потребить мышцами — таких цифр тоже никто ещё не зафиксировал. Обычно потребляют где-то 6 л/мин, ну — 6,5 л/мин, цифры в 7 л/мин кислорода почти не появляется.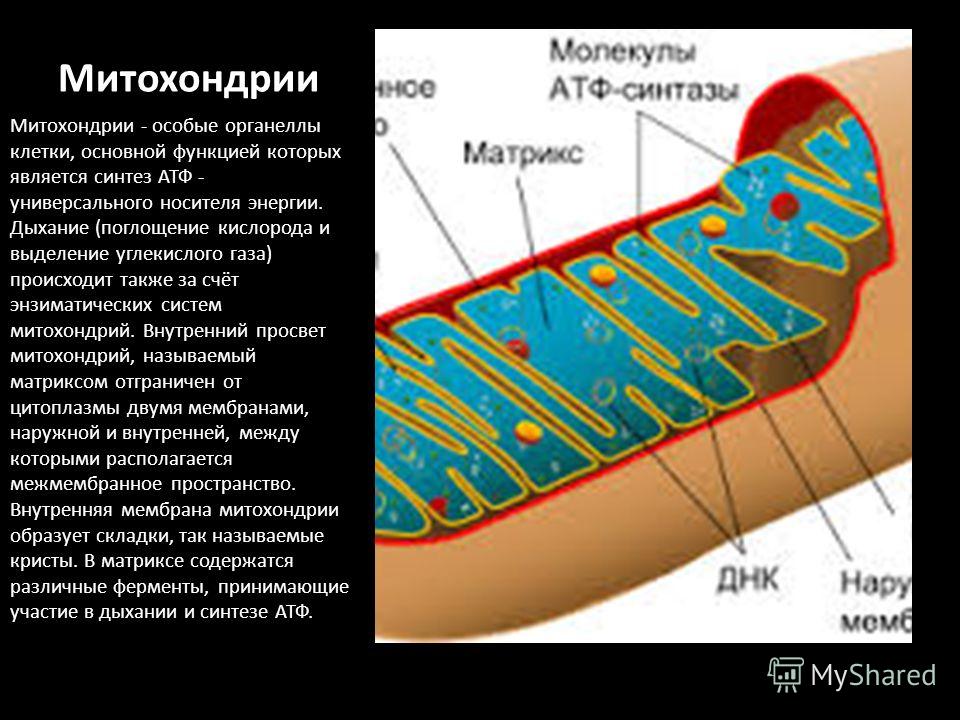
Тестирование уровней потребления кислорода поможет построить планы тренировок
Поскольку работоспособность может лимитировать либо одно, либо другое, то для того, чтобы разобраться с тем, чего не хватает конкретному спортсмену — его надо обязательное тестировать. Например, мы начинаем тестировать лыжников на уровне сборной команды, и получаем очень печальные цифры. Фиксируем показатели неоднократного победителя крупных российских марафонов (спортсмен каждый год в «десятке» на чемпионатах России), и видим: мышцы ног потребляют кислорода только 3,5 л/мин на уровне АнП — это результат порядка 1 разряда в велосипедном спорте. А лыжник должен потреблять ногами столько же, сколько велосипедист МСМК, причем это абсолютная цифра, не на килограмм веса. (В велосипедном спорте это не принципиально, там больше важно, что приходится на лобовую площадь.)
Спрашивается, а какое у него сердце? Если взять график ступенчатого теста, то на начальном участке, когда рекрутируются только ОМВ, наблюдается некая прямая между пульсом и мощностью. Потом эта кривая зависимости (потом кривая получается) начинает как-то изменяться. И, как правило, происходит увеличение темпа прироста пульса. Если продолжить начальный отрезок линии дальше, и вывести на пульс 190, то можно предсказать, что бы было с этим человеком, если бы он вышел на пульс 190, и при этом у него были бы только ОМВ. И тогда мы определили бы потенциальные возможности сердца по доставке кислорода к мышцам. (Подробнее об этом можно прочитать в следующем номере в разделе, посвященном интерпретации данных ступенчатого теста).
Потом эта кривая зависимости (потом кривая получается) начинает как-то изменяться. И, как правило, происходит увеличение темпа прироста пульса. Если продолжить начальный отрезок линии дальше, и вывести на пульс 190, то можно предсказать, что бы было с этим человеком, если бы он вышел на пульс 190, и при этом у него были бы только ОМВ. И тогда мы определили бы потенциальные возможности сердца по доставке кислорода к мышцам. (Подробнее об этом можно прочитать в следующем номере в разделе, посвященном интерпретации данных ступенчатого теста).
Так вот, потенциальная производительность сердца оказывается у него 7 л/мин. Это означает, что наш спортсмен имеет прекрасное сердце, огромное сердце, его тренировать специально не надо, а мышцы, прежде всего ног, — очень слабые, они в очень плохом состоянии, их надо готовить, чтобы они соответствовали нормативам международного класса.
Чтобы этот лыжник показал хорошие результаты, ему надо где-то 4,5 л/мин потреблять ногами. С показателем 4,5 л/мин он бы в сборной уже устойчиво стоял. При этом пульс у него при потреблении кислорода 4,5 л/мин должен быть не 190 уд/мин, а 150, потому что должен быть запас, на котором руки будут работать. Хорошо, предположим, мы с ним в тесте получаем 4,5 л/мин на пульсе 150 уд/мин, и после этого начинается закисление, и он отказывается от работы. Тогда мы говорим, что ноги у него в хорошем состоянии (4,5 л/мин для лыжника вполне достаточно). Потом начинаем тестировать руки, и оказывается, что руки у него потребляют где-то 1,5 л/мин, больше не будут потреблять (это из нашего опыта известно). Он потребляет руками 1,5 л/мин, мы прибавляем их к 4,5 л/мин ног, и получаем потребление кислорода равное 6 л/мин. Затем делим на его вес 70 кг и получаем 85 мл/кг/мин — это уровень олимпийских достижений.
При этом пульс у него при потреблении кислорода 4,5 л/мин должен быть не 190 уд/мин, а 150, потому что должен быть запас, на котором руки будут работать. Хорошо, предположим, мы с ним в тесте получаем 4,5 л/мин на пульсе 150 уд/мин, и после этого начинается закисление, и он отказывается от работы. Тогда мы говорим, что ноги у него в хорошем состоянии (4,5 л/мин для лыжника вполне достаточно). Потом начинаем тестировать руки, и оказывается, что руки у него потребляют где-то 1,5 л/мин, больше не будут потреблять (это из нашего опыта известно). Он потребляет руками 1,5 л/мин, мы прибавляем их к 4,5 л/мин ног, и получаем потребление кислорода равное 6 л/мин. Затем делим на его вес 70 кг и получаем 85 мл/кг/мин — это уровень олимпийских достижений.
Дальше разбираемся, что с ним нужно делать, чтобы достичь таких показателей. Так вот, первый вывод: поскольку сердце у него большое, и может перекачать кислорода 7 л/мин, то этому человеку не надо делать вкатывание. Под вкатыванием понимаются объёмные длительные тренировки продолжительностью от 3 до 6-8 часов в день на относительно низком пульсе (100-150 уд/мин, близко к 120).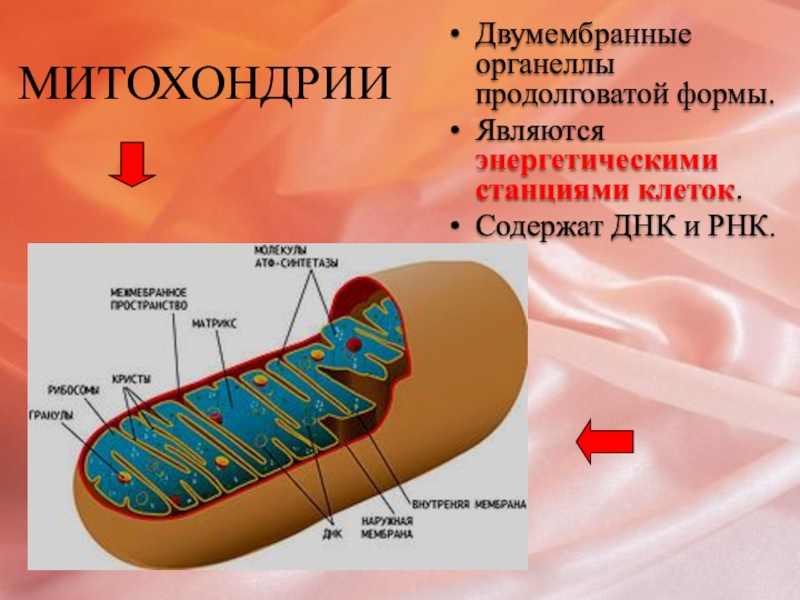 Если человек будет по 8 часов в день кататься при таком пульсе, то сердце начнет дилятировать (расширяться) и может значительно увеличиться в объеме. А этому человеку нужно заниматься в первую очередь мышцами ног — именно они ограничивают его возможности.
Если человек будет по 8 часов в день кататься при таком пульсе, то сердце начнет дилятировать (расширяться) и может значительно увеличиться в объеме. А этому человеку нужно заниматься в первую очередь мышцами ног — именно они ограничивают его возможности.
А у другого может оказаться наоборот. Вот вам следующий пример: еще один молодой перспективный лыжник, мы его тестируем, у него картина такая: пульс 190 уд/мин и 4,5 л/мин потребляет ногами, но пульс-то — 190. Всё, ему руки нельзя добавлять, он на пределе, сердце маленькое, слабое. Это было как раз в 2000 году, когда он ряд гонок выиграл и, как говорят, «капнул». Его больше в сборную брать не стали — сердце не держит. Никто же этого не знает, но чувствуют — спортсмен начинает проигрывать, не держит тренировочных нагрузок. Сердце маленькое. Наконец, дали ему отдохнуть, выбросили все объёмные нагрузки, оставили только интенсивные, спринтерского характера. Сердце постепенно вылечилось, за 4-5 месяцев стало нормальным, стало свои 8 л/мин качать, вместо 4,5 л/мин.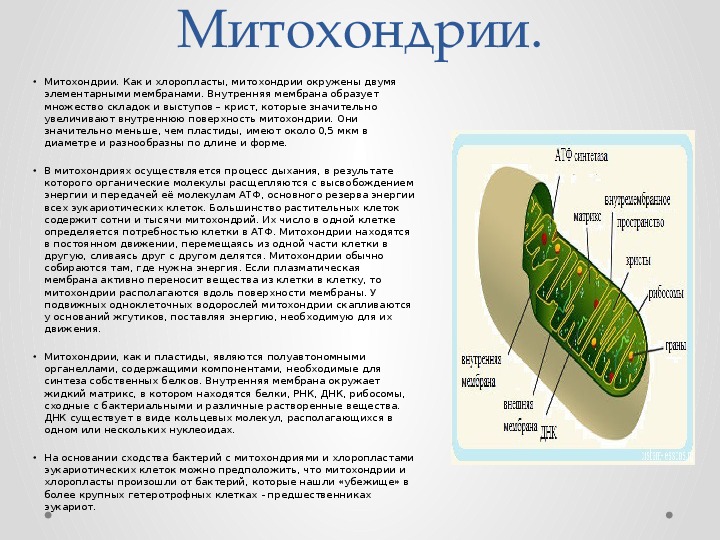 Потребление кислорода в руках добавили, чуть ли не в 2 раза, а ноги у него и так хорошие. Он свои 4,5 л/мин ногами как потреблял, так и потребляет, но на пульсе не 190 уд/мин, а 160, потом руки еще добавляет, и он выходит на пульс 190, на этом пульсе можно бежать 10 км. У него был явный недостаток сердца, но причина не в том, что сердце плохое, ему надо было просто дать восстановиться, чтобы прекратились дистрофические явления, и он вернулся в нормальное состояние.
Потребление кислорода в руках добавили, чуть ли не в 2 раза, а ноги у него и так хорошие. Он свои 4,5 л/мин ногами как потреблял, так и потребляет, но на пульсе не 190 уд/мин, а 160, потом руки еще добавляет, и он выходит на пульс 190, на этом пульсе можно бежать 10 км. У него был явный недостаток сердца, но причина не в том, что сердце плохое, ему надо было просто дать восстановиться, чтобы прекратились дистрофические явления, и он вернулся в нормальное состояние.
Заключение
Хочется добавить о роли интуиции и знания. Про роль знания особо говорить не станем – этому посвящена вся статья. Что касается интуиции, приведем выдержку из книги Виктора Николаевича «Подготовка бегуна на средние дистанции»:
«Принцип интуиции. Каждый спортсмен должен опираться в тренировке не только на правила, но и на интуицию, поскольку имеются индивидуальные особенности адаптационных реакций».
Принцип интуиции можно переформулировать иначе – «Природа «умнее» любого ученого».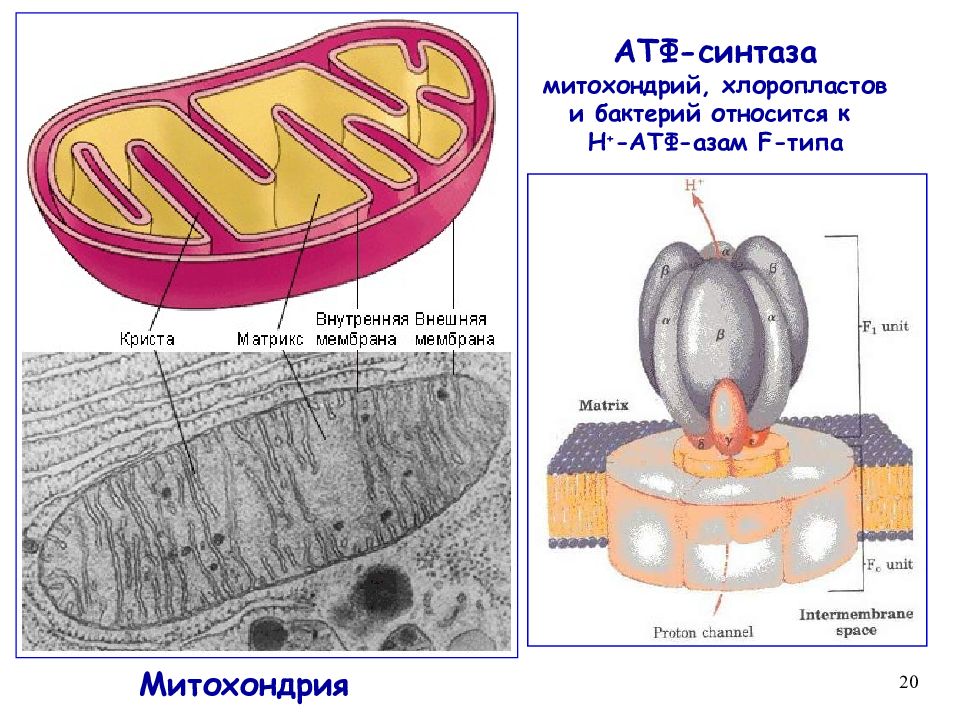 Поэтому можно планировать тренировочный процесс, но при его реализации спортсмен должен «прислушиваться» к своим ощущениям, сопоставлять их с ожиданиями своими и тренера. Разумеется, необходимо как можно чаще контролировать состояние готовности различных систем и органов. Эта информация, вместе со знаниями, является основой продуктивной интуиции, творческих озарений в построении тренировочного процесса. В связи с этим действительно можно принять утверждение «Знание слепо без интуиции», когда идет работа с конкретным спортсменом, когда приходится вводить коррекцию в тренировочный процесс.
Поэтому можно планировать тренировочный процесс, но при его реализации спортсмен должен «прислушиваться» к своим ощущениям, сопоставлять их с ожиданиями своими и тренера. Разумеется, необходимо как можно чаще контролировать состояние готовности различных систем и органов. Эта информация, вместе со знаниями, является основой продуктивной интуиции, творческих озарений в построении тренировочного процесса. В связи с этим действительно можно принять утверждение «Знание слепо без интуиции», когда идет работа с конкретным спортсменом, когда приходится вводить коррекцию в тренировочный процесс.
Тяжёлый изнурительный бег вверх по горной тропе, использовавшийся Бьорном Дали и Вегардом Ульвангом, чем-то напоминает бег по холмам в Новозеландии учеников Лидьярда.
При подготовке статьи использованы разработки В. Н. Селуянова
Обсудить на форуме
комментарии к статье
Пока нет комментариев
Methods to Assess Subcellular Compartments of Muscle in C.
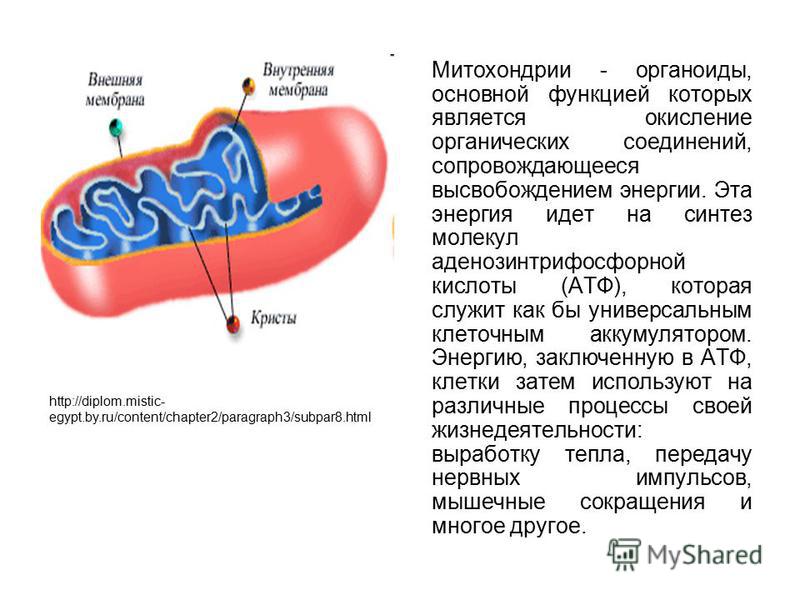 elegans
elegansЭти методы позволяют разведку мышцы от macrophysiology движения для идентификации субклеточных дефекты, которые достаточно, чтобы вызвать дефект движения. В сочетании эти методы позволяют детальный анализ мышцы. Понимание получила позволяет дальнейшее тестирование мышц-ориентированных гипотез, относящихся к общей физиологии, патофизиологии и старения.
Движение Анализы
Дефект движение указывает на нарушение нормальной нервно-мышечной функции. Это могут быть специфическими для нервов или мышц, или он может быть общим между несколькими тканей. Самые трудные этапы комплектующих анализов движения выбираете червей, которые являются репрезентативными для населения и / или лечения, а также обеспечение того, чтобы экспериментатор не травмировать животное при переходе на М9. Важно иметь большой размер выборки, чтобы получить представительную и точную оценку движения. Когда анализируя высоко пероetrant эффекты, например, как часто можно увидеть в мутантов, размер выборки из десяти животных каждый оценивается для движения в десять раз достаточно найти статистически значимых различий в сравнении дикого типа. Тем не менее, простота анализов движения и легкости культивирования C. Элеганс означает, что сотни червей быстро можно оценить с тем чтобы обеспечить количественно надежные результаты. Когда анализируя эффекты, которые появляются присутствует только в части популяции важно, чтобы определить, какая часть населения по-видимому, будут затронуты, а также от тяжести дефекта в наиболее пораженных животных. В таких ситуациях большее число животных должна быть оценена, чтобы обеспечить минимальные данные для части населения пострадавших. Часто оба препарата и РНК-интерференции лечения будет производить тяжелые дефекты движения в небольшой части населения. В некоторых случаях увеличение дозы лечения и или способа доставки будет увеличивать долю AFFected животные и кривые зависимости ответа от дозы бег может быть полезен при определении оптимальной концентрации лекарственного средства или РНК-интерференции. Второе ограничение этого движения анализе является то, что черви манипулировать, чтобы оценить движение.
Тем не менее, простота анализов движения и легкости культивирования C. Элеганс означает, что сотни червей быстро можно оценить с тем чтобы обеспечить количественно надежные результаты. Когда анализируя эффекты, которые появляются присутствует только в части популяции важно, чтобы определить, какая часть населения по-видимому, будут затронуты, а также от тяжести дефекта в наиболее пораженных животных. В таких ситуациях большее число животных должна быть оценена, чтобы обеспечить минимальные данные для части населения пострадавших. Часто оба препарата и РНК-интерференции лечения будет производить тяжелые дефекты движения в небольшой части населения. В некоторых случаях увеличение дозы лечения и или способа доставки будет увеличивать долю AFFected животные и кривые зависимости ответа от дозы бег может быть полезен при определении оптимальной концентрации лекарственного средства или РНК-интерференции. Второе ограничение этого движения анализе является то, что черви манипулировать, чтобы оценить движение. Таким образом, черви и стимулируется и подвергают какой-то степени осмотического стресса при получении в жидкость. Степень осмотического стресса может быть ограничен с помощью M9 или BU буфера 19, в отличие от воды. Некоторые из этих ограничений смягчаются путем измерения привычный движение на пластинах с использованием коммерчески доступных системы слежения 34 или бесплатные плагины для ImageJ 35. Измерение скорости движения животного могут предоставить важную информацию о здоровье в целом мышц, в том числе изменения в функции, которые можно измерить на протяжении всей жизни и не-инвазивность этого метода является ключевым для потенциальных пожизненных исследований функции мышц.
Таким образом, черви и стимулируется и подвергают какой-то степени осмотического стресса при получении в жидкость. Степень осмотического стресса может быть ограничен с помощью M9 или BU буфера 19, в отличие от воды. Некоторые из этих ограничений смягчаются путем измерения привычный движение на пластинах с использованием коммерчески доступных системы слежения 34 или бесплатные плагины для ImageJ 35. Измерение скорости движения животного могут предоставить важную информацию о здоровье в целом мышц, в том числе изменения в функции, которые можно измерить на протяжении всей жизни и не-инвазивность этого метода является ключевым для потенциальных пожизненных исследований функции мышц.
Визуализация сократительного аппарата
Штамм червь используется здесь для структуры изображения сократительного аппарата был PJ727 36 выражающее миозина GFP слитый белок, который ограничен к саркомерах в стенки тела мышц. Использование этого штамма позволяет визуализировать структуры саркомера в живых животных. Аналогично движению анализа, описанного выше, ключевым преимуществом использования GFP помечены саркомеры оценить структуру саркомера является то, что метод является относительно неинвазивным и, следовательно, может быть использован для перспективно следовать структуру саркомера в отдельных животных на протяжении всей жизни. Наибольшую трудность этого метода является то, что черви, которые изображаемого живы и поэтому движется, что делает его трудно получить хорошо сфокусированные изображения. Несколько методов могут быть использованы, чтобы уменьшить движение и сделать более простым визуализации; они включают обезболивающее или парализует червей и иммобилизации через давление, всасывания или использованием высокой решения вязкости. Каждый из этих методов immobilizция имеет тот недостаток, что он может сам изменять нервно-мышечную функцию. Таким образом, крайне важно, чтобы включать в себя соответствующие необработанными контрольными в анализе. Он также может быть целесообразно использовать по крайней мере два независимых методов иммобилизации, чтобы подтвердить результаты не из-за проблем с методом иммобилизации, в зависимости от того, как широко используемый метод иммобилизации выбранные для изучаемого вопроса.
Аналогично движению анализа, описанного выше, ключевым преимуществом использования GFP помечены саркомеры оценить структуру саркомера является то, что метод является относительно неинвазивным и, следовательно, может быть использован для перспективно следовать структуру саркомера в отдельных животных на протяжении всей жизни. Наибольшую трудность этого метода является то, что черви, которые изображаемого живы и поэтому движется, что делает его трудно получить хорошо сфокусированные изображения. Несколько методов могут быть использованы, чтобы уменьшить движение и сделать более простым визуализации; они включают обезболивающее или парализует червей и иммобилизации через давление, всасывания или использованием высокой решения вязкости. Каждый из этих методов immobilizция имеет тот недостаток, что он может сам изменять нервно-мышечную функцию. Таким образом, крайне важно, чтобы включать в себя соответствующие необработанными контрольными в анализе. Он также может быть целесообразно использовать по крайней мере два независимых методов иммобилизации, чтобы подтвердить результаты не из-за проблем с методом иммобилизации, в зависимости от того, как широко используемый метод иммобилизации выбранные для изучаемого вопроса. Здесь мы использовали простой давление от покровного стекла на червя, чтобы вызвать пониженное движение. После размещения покровное, жидкость под покровным испарится и покровное будет, следовательно, начинают производить больше давление на червей, как правило, это занимает 2-5 минуты, чтобы произвести достаточное снижение движение, чтобы позволить легкий изображений.
Здесь мы использовали простой давление от покровного стекла на червя, чтобы вызвать пониженное движение. После размещения покровное, жидкость под покровным испарится и покровное будет, следовательно, начинают производить больше давление на червей, как правило, это занимает 2-5 минуты, чтобы произвести достаточное снижение движение, чтобы позволить легкий изображений. Важно следить за червей тесно после покровное была введена как давление в конечном итоге увеличить достаточно, чтобы вызвать червей лопнуть от избыточного давления, как правило, 10-15 минут после сотрудничестваverslip дополнение. Дополнительная буфера могут быть введены с помощью наконечника пипетки вокруг краев покровного стекла, чтобы избежать травм на раздавливание червей. Окрашивание ногтей может быть использован для герметизации краев покровным стеклом и предотвратить испарение, хотя это предотвращает извлечение животных из-под покровным стеклом, при желании. Точно так же, использование силиконовых шариков в растворе может помочь уменьшить количество давлением покровного места на червей.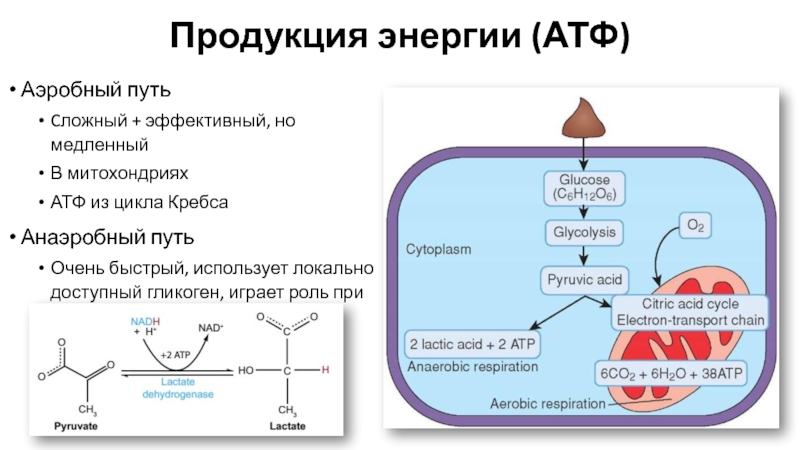
Так же, как с оценкой за дефекта движения, важно рассмотреть пенетрантность эффекта. Пенетрантность можно считать высокой, если 80-100% животных отображения фенотип на NGM пластины, частичное если 21-79% и низкой, если ≤20% от дисплея населения влияет. Для сильно проникающих эффектов, меньшие размеры выборки могут быть использованы для производства значительных количественных данных о саркомера дефектов. В тех случаях, когда эффект имеет низкую пенетрантность, выбор животных, что четкое отображение дефект движение в ответ на лекарстваили лечение RNAi, могут быть полезны в опробования саркомера дефекты у животных, наиболее пострадавших от лечения. Тем не менее, это не обязательно так, что степень эффективности лечения на движения и субклеточном структуре такой же. Таким образом, может быть желательно, чтобы исследовать степень дефектов, присутствующих в большой популяции, например 100 животных. Это дает возможность понимания распределения дефектов по всей популяции. Он также может быть желательно, чтобы изучить степень дефекта в наиболее пострадавших животных и / или изучить корреляцию между степенью саркомера дефекта и степени дефекта движения.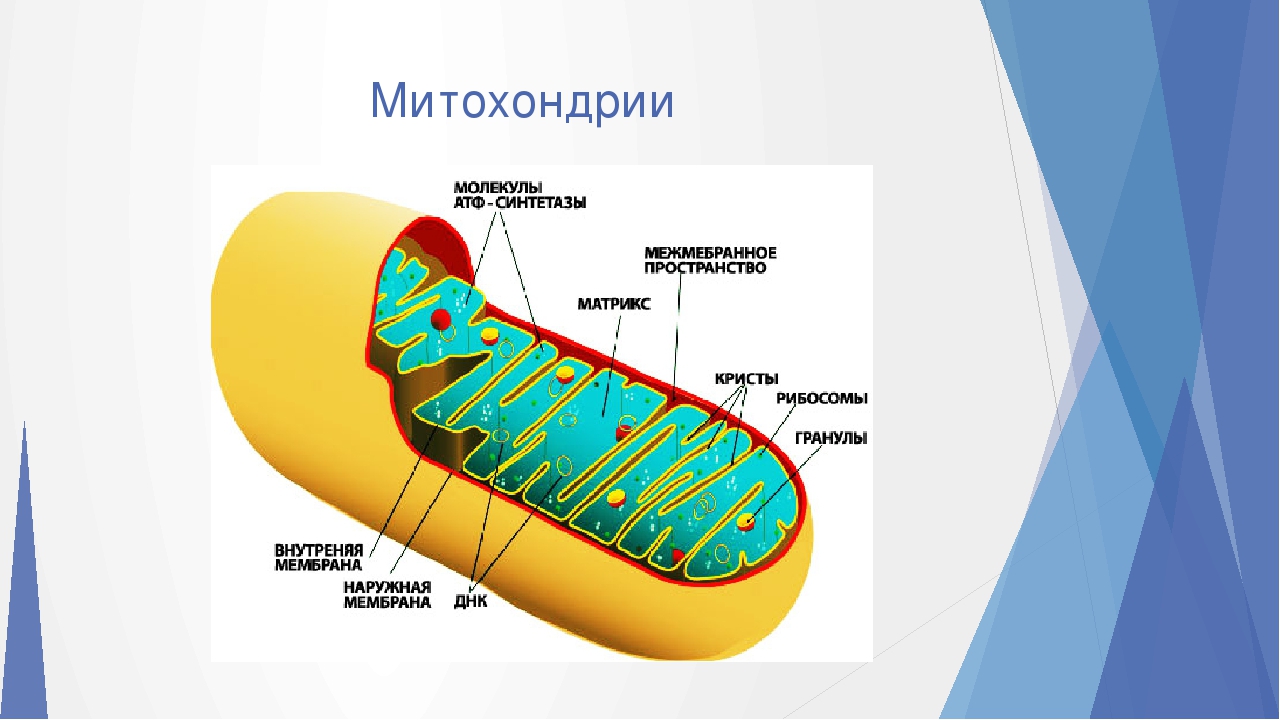 Потенциальные трудности в сторону, этот метод быстрее и проще, чем другие методы оценки структуры саркомера. Эта скорость и легкость, однако, при условии ключевого ограничения, саркомеры слитые белки, содержащие GFP не совсем нормально 37 и, следовательно, оценка разрушенных саркомеров должен быть подтвержден с использованием дополнительных более трудоемких методов.Эти методы включают в себя использование поляризованного света 38, фаллоидином окрашивания 39 и непрямой иммунофлюоресценции 40. Из этих методов, только поляризованный свет является относительно неинвазивным; другие методы требуют фиксации и, следовательно, не могут быть использованы в перспективных исследований отдельных животных, а они могут быть использованы только для подтверждения, пересекают сечении, результаты таких исследований.
Потенциальные трудности в сторону, этот метод быстрее и проще, чем другие методы оценки структуры саркомера. Эта скорость и легкость, однако, при условии ключевого ограничения, саркомеры слитые белки, содержащие GFP не совсем нормально 37 и, следовательно, оценка разрушенных саркомеров должен быть подтвержден с использованием дополнительных более трудоемких методов.Эти методы включают в себя использование поляризованного света 38, фаллоидином окрашивания 39 и непрямой иммунофлюоресценции 40. Из этих методов, только поляризованный свет является относительно неинвазивным; другие методы требуют фиксации и, следовательно, не могут быть использованы в перспективных исследований отдельных животных, а они могут быть использованы только для подтверждения, пересекают сечении, результаты таких исследований.
Визуализация митохондриальных Networks
Штамм червь используется для митохондриальных экспериментов было визуализации CB5600 7, который выражает GFP слитый белок локализован в митохондриях и GFP β-галактозидазы слитого белка, локализованного в ядрах, как в теле стеновых мышц. Как с использованием PJ727 для визуализации саркомерах, использование данного штамма позволяет визуализировать митохондриальной структуры сети в живых животных. Таким образом, этот штамм может быть использован для оценки динамических изменений, которые происходят, например, снижается mitochondrial слияние и / или увеличение митохондрий деления 41. Также как и для визуализации саркомерах, самая большая трудность при этом способе является то, что черви изображаемого живы и перемещения, что делает его трудно получить хорошо сфокусированные изображения. В то время как несколько методов, как описано более подробно выше, могут быть использованы, чтобы уменьшить движение, митохондрии появляются особенно чувствительны к вмешательств, которые индуцируют уменьшенную движение. Например, наиболее часто используемый анестетики целевые компоненты дыхательной цепи митохондрий и, следовательно, должны использоваться с осторожностью в исследованиях нормальной митохондриальной структуры и функции.
Как с использованием PJ727 для визуализации саркомерах, использование данного штамма позволяет визуализировать митохондриальной структуры сети в живых животных. Таким образом, этот штамм может быть использован для оценки динамических изменений, которые происходят, например, снижается mitochondrial слияние и / или увеличение митохондрий деления 41. Также как и для визуализации саркомерах, самая большая трудность при этом способе является то, что черви изображаемого живы и перемещения, что делает его трудно получить хорошо сфокусированные изображения. В то время как несколько методов, как описано более подробно выше, могут быть использованы, чтобы уменьшить движение, митохондрии появляются особенно чувствительны к вмешательств, которые индуцируют уменьшенную движение. Например, наиболее часто используемый анестетики целевые компоненты дыхательной цепи митохондрий и, следовательно, должны использоваться с осторожностью в исследованиях нормальной митохондриальной структуры и функции.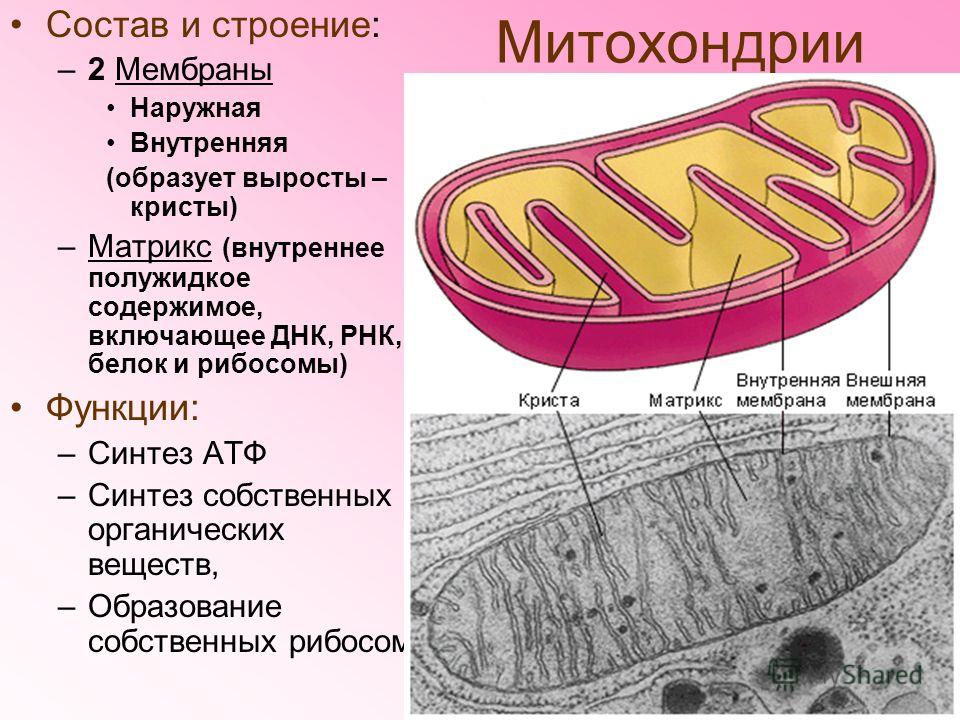 Точно так же большинство паралитические агенты должны использоваться с осторожностью в исследованиях нормальной митохондриальной структуры и функции, как большинство паралитические агенты вызывают меньше сигнал проходит через нервно-мышечного соединения и функциональной денервации мышцы достаточно, чтобы вызвать митохондриальную фрагментации. Эксперименты в этом исследовании использовали давление, чтобы иммобилизовать животноеS, но другие успешно использован левамизол 42, хотя это имеет важное значение для изображения животных сразу.
Точно так же большинство паралитические агенты должны использоваться с осторожностью в исследованиях нормальной митохондриальной структуры и функции, как большинство паралитические агенты вызывают меньше сигнал проходит через нервно-мышечного соединения и функциональной денервации мышцы достаточно, чтобы вызвать митохондриальную фрагментации. Эксперименты в этом исследовании использовали давление, чтобы иммобилизовать животноеS, но другие успешно использован левамизол 42, хотя это имеет важное значение для изображения животных сразу.
Наконец, различные бактериальные загрязняющие вещества в культурах, например Б. Сенная, может вызвать митохондриальную фрагментации; Поэтому очень важно, чтобы включить соответствующие необработанные управления в анализе. Для сильно проникающих эффектов, меньшие размеры выборки могут быть использованы для производства значительные количественные данные о сетевых митохондриальных дефектов. Тем не менее, из-за распространенности незначительными дефектами в митохондриальной сети, это небольшое количество животных, вероятно, будет в два раза больше количества, необходимого для оценки структуры саркомера. В тех случаях, когда эффект имеет низкую пенетрантность, выбор животных четко отображать дефект движение в ответ на лекарства или лечения RNAi, могут быть полезны в опробования митохондриальные дефекты у животных, наиболее пострадавших от лечения. Тем не менее, как упоминалось выше, это не всегда так, чтоСтепень эффекта лечения на движения и субклеточном структуре такой же. Таким образом, может быть желательно, чтобы изучить степень дефектов, имеющихся в большой популяции, например 150-200 животных, а также степень нарушения в наиболее сильно пострадавших животных и наличие или отсутствие степени нарушения со степенью дефект движение. Другие штаммы с флуоресцентно меченый митохондрий может быть использована, чтобы подтвердить результаты, полученные в CB5600, однако, использование различных флуоресцентных белков могут влиять на базовой сети митохондрий. Например, использование Томма-20 RFP слитого белка для визуализации митохондрий появляется, чтобы вызвать увеличение базовой митохондриальной фрагментации по сравнению с использованием митохондриально локализованного GFP 17.
В тех случаях, когда эффект имеет низкую пенетрантность, выбор животных четко отображать дефект движение в ответ на лекарства или лечения RNAi, могут быть полезны в опробования митохондриальные дефекты у животных, наиболее пострадавших от лечения. Тем не менее, как упоминалось выше, это не всегда так, чтоСтепень эффекта лечения на движения и субклеточном структуре такой же. Таким образом, может быть желательно, чтобы изучить степень дефектов, имеющихся в большой популяции, например 150-200 животных, а также степень нарушения в наиболее сильно пострадавших животных и наличие или отсутствие степени нарушения со степенью дефект движение. Другие штаммы с флуоресцентно меченый митохондрий может быть использована, чтобы подтвердить результаты, полученные в CB5600, однако, использование различных флуоресцентных белков могут влиять на базовой сети митохондрий. Например, использование Томма-20 RFP слитого белка для визуализации митохондрий появляется, чтобы вызвать увеличение базовой митохондриальной фрагментации по сравнению с использованием митохондриально локализованного GFP 17. В то время как другие методы, такие как иммунофлюоресценции и электронной микроскопии может быть использован для оценки митохондриальной структуры, они не позволяют оценку в естественных условиях митохондриальных динамики, потому что шаг крепление требуетd. Другие методы, такие как использование митохондрии локализованных красителей можно использовать без фиксации и, следовательно, также могут быть использованы вместо использования митохондриально локализованного GFP. Как обсуждается в следующем разделе, использование таких красителей позволяет сырой оценку в естественных условиях функции митохондрий.
В то время как другие методы, такие как иммунофлюоресценции и электронной микроскопии может быть использован для оценки митохондриальной структуры, они не позволяют оценку в естественных условиях митохондриальных динамики, потому что шаг крепление требуетd. Другие методы, такие как использование митохондрии локализованных красителей можно использовать без фиксации и, следовательно, также могут быть использованы вместо использования митохондриально локализованного GFP. Как обсуждается в следующем разделе, использование таких красителей позволяет сырой оценку в естественных условиях функции митохондрий.
Оценка митохондриальной мембране потенциала в Vivo в С. Элеганс
Разнообразие красителей доступны для маркировки митохондрий 28. Эти красители обеспечивают альтернативный метод визуализации митохондрий структуру сети в живых C. Элеганс. Многие из этих красителей также позволить сырой оценку митохондриальной функциональности в естественных условиях, так как они накапливаются в митохондриях, основанных на митохондриальной мембранного потенциала. Здесь мы использовали C 32 H 32 Cl 2 N 2 O, которая попадает в организм черезпищеварительного тракта, при этом некоторые дополнительно ввод червя через кутикулы из-за проницаемости, предоставляемой растворения в 0,5% ДМСО. После вступления в клетке C 32 H 32 Cl 2 N 2 O окисляется и поглощенных по митохондрии, где он связывается серосодержащих аминокислот (например, метионина и цистеина). Образование этих красителей комплексов пептид-C означает 32 H 32 Cl 2 N 2 O остается в митохондриях раз меченых 28. Основной трудностью при использовании красителей митохондриальных является то, что они будут маркировать все митохондрии в животном. Поэтому мы провели маркировку в CB5600, и того же штамма, описанного выше для оценки мышечной митохондриальной структуры сети. Используя красную митохондриальной краситель, штамм червей, выражающих GFP в мышечной митохондрий, и тройной фильтр, это относительно просто изображение только митохондрии в мышцах, находя митохондрии, которые являются уплотнительныеДиапазон по колокализации красным красителем и GFP меченных митохондрий.
Здесь мы использовали C 32 H 32 Cl 2 N 2 O, которая попадает в организм черезпищеварительного тракта, при этом некоторые дополнительно ввод червя через кутикулы из-за проницаемости, предоставляемой растворения в 0,5% ДМСО. После вступления в клетке C 32 H 32 Cl 2 N 2 O окисляется и поглощенных по митохондрии, где он связывается серосодержащих аминокислот (например, метионина и цистеина). Образование этих красителей комплексов пептид-C означает 32 H 32 Cl 2 N 2 O остается в митохондриях раз меченых 28. Основной трудностью при использовании красителей митохондриальных является то, что они будут маркировать все митохондрии в животном. Поэтому мы провели маркировку в CB5600, и того же штамма, описанного выше для оценки мышечной митохондриальной структуры сети. Используя красную митохондриальной краситель, штамм червей, выражающих GFP в мышечной митохондрий, и тройной фильтр, это относительно просто изображение только митохондрии в мышцах, находя митохондрии, которые являются уплотнительныеДиапазон по колокализации красным красителем и GFP меченных митохондрий. Самым сложным этапом в выполнении этой колокализации подход является визуализация потому краситель фото выгоревшие в течение нескольких секунд при высокой интенсивности флуоресцентного света. Этот эффект может быть ограничен, сохраняя флуоресценцию на малой мощности до тех пор, экспериментатор не будет готов к изображению, а затем регулировки интенсивности флуоресценции, соответственно. После того, как один испытывают с использованием красителей и другой подход заключается в использовании конфокальной микроскопии с Z-стеки в образ только митохондрии в правой ткани; митохондриальные сети в мышце совершенно различны по сравнению с другими тканями. К сожалению, неспособность C 32 H 32 Cl 2 N 2 O, чтобы оставить митохондрии 28 означает, что, в отличие от методики саркомера, и митохондриальной изображений обсуждалось выше, он не может быть использован, чтобы перспективно следовать потерю функции митохондрий со временем, если C 32 H 32 Cl 2 N 2 Oдобавляется в дискретные моменты времени, чтобы отделить популяции животных.
Самым сложным этапом в выполнении этой колокализации подход является визуализация потому краситель фото выгоревшие в течение нескольких секунд при высокой интенсивности флуоресцентного света. Этот эффект может быть ограничен, сохраняя флуоресценцию на малой мощности до тех пор, экспериментатор не будет готов к изображению, а затем регулировки интенсивности флуоресценции, соответственно. После того, как один испытывают с использованием красителей и другой подход заключается в использовании конфокальной микроскопии с Z-стеки в образ только митохондрии в правой ткани; митохондриальные сети в мышце совершенно различны по сравнению с другими тканями. К сожалению, неспособность C 32 H 32 Cl 2 N 2 O, чтобы оставить митохондрии 28 означает, что, в отличие от методики саркомера, и митохондриальной изображений обсуждалось выше, он не может быть использован, чтобы перспективно следовать потерю функции митохондрий со временем, если C 32 H 32 Cl 2 N 2 Oдобавляется в дискретные моменты времени, чтобы отделить популяции животных.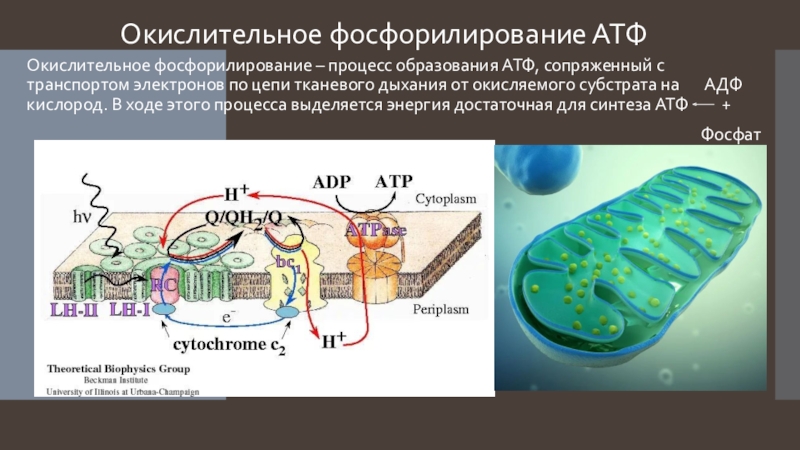 Это ограничение можно преодолеть, используя красители, такие как JC-10, что делать оставить митохондрии от распада мембранного потенциала 43. Митохондрии также могут быть выделены из С. Элеганс 30 для последующих биохимических анализов. Такая добыча позволяет количественно оценить митохондриальной мембранного потенциала путем количественного определения скорости поглощения липофильных катионный краситель с помощью проточной цитометрии или люминесцентной ридере 44. Установив нарушенную митохондриальный мембранный потенциал, можно также определить, если потеря протонного градиента приводит к потере производства АТФ с помощью чувствительного bioluminometric люциферазы на основе метода 45.
Это ограничение можно преодолеть, используя красители, такие как JC-10, что делать оставить митохондрии от распада мембранного потенциала 43. Митохондрии также могут быть выделены из С. Элеганс 30 для последующих биохимических анализов. Такая добыча позволяет количественно оценить митохондриальной мембранного потенциала путем количественного определения скорости поглощения липофильных катионный краситель с помощью проточной цитометрии или люминесцентной ридере 44. Установив нарушенную митохондриальный мембранный потенциал, можно также определить, если потеря протонного градиента приводит к потере производства АТФ с помощью чувствительного bioluminometric люциферазы на основе метода 45.
Оценивая белка деградации в С. Элеганс
Штамм червь используется здесь для оценки деградации мышечных белков был PD55, который содержит мышц конкретный βгалактозидазу что постоянно синтезируется в ходе развития до зрелого возраста не будет достигнут и который остается стабильным в цитозоле для следующего 72-96 часов 19.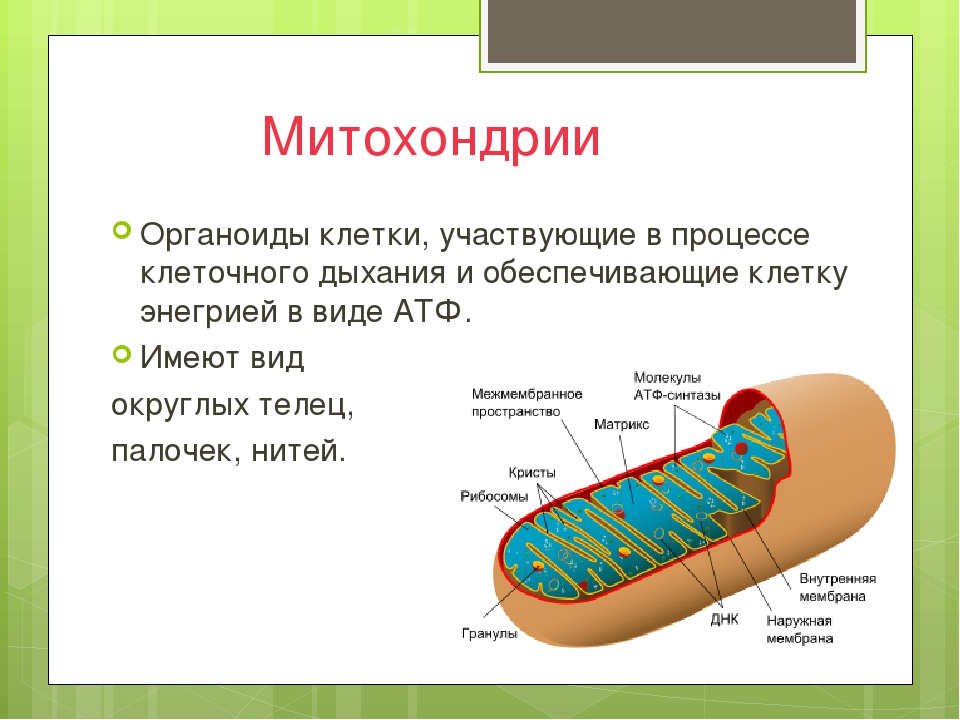 Таким образом, измерение уровня β-галактозидазы в процессе разработки преимущественно указывает на воздействие мероприятий на синтез из миозина промоутера в то время как потеря β-галактозидазы в зрелом возрасте показывает вмешательство вызвало увеличилось деградации цитозольный белка 19. Ключевым шагом в оценке β-галактозидазы является обеспечение эффективного высыхания. Несоблюдение эффективно пересушивает животных приводит к плохому предоставления X-гал субстрата и, следовательно, плохой синего цвета в тело-стеновых мышц животных. Для обеспечения хорошего высыхания печать на эксикаторе должны быть проверены и образец должен быть проверен на 5-10-минутными интервалами на протяжении высыхания для оценки прогресса (т.е. образца 20 мкл следует высохли в пределах 10 минут, а оставшиеся 20 минявляется обеспечение всестороннего высыхания). Β-галактозидазы анализ является анализ активности Поэтому соответствующий контроль имеет важное значение. Кроме того, снизилась окрашивание взрослых в состояние обращение относительно контроля не доказывает, что деградация происходит; а это указывает на снижение ферментативной активности в ответ на лечение.
Таким образом, измерение уровня β-галактозидазы в процессе разработки преимущественно указывает на воздействие мероприятий на синтез из миозина промоутера в то время как потеря β-галактозидазы в зрелом возрасте показывает вмешательство вызвало увеличилось деградации цитозольный белка 19. Ключевым шагом в оценке β-галактозидазы является обеспечение эффективного высыхания. Несоблюдение эффективно пересушивает животных приводит к плохому предоставления X-гал субстрата и, следовательно, плохой синего цвета в тело-стеновых мышц животных. Для обеспечения хорошего высыхания печать на эксикаторе должны быть проверены и образец должен быть проверен на 5-10-минутными интервалами на протяжении высыхания для оценки прогресса (т.е. образца 20 мкл следует высохли в пределах 10 минут, а оставшиеся 20 минявляется обеспечение всестороннего высыхания). Β-галактозидазы анализ является анализ активности Поэтому соответствующий контроль имеет важное значение. Кроме того, снизилась окрашивание взрослых в состояние обращение относительно контроля не доказывает, что деградация происходит; а это указывает на снижение ферментативной активности в ответ на лечение.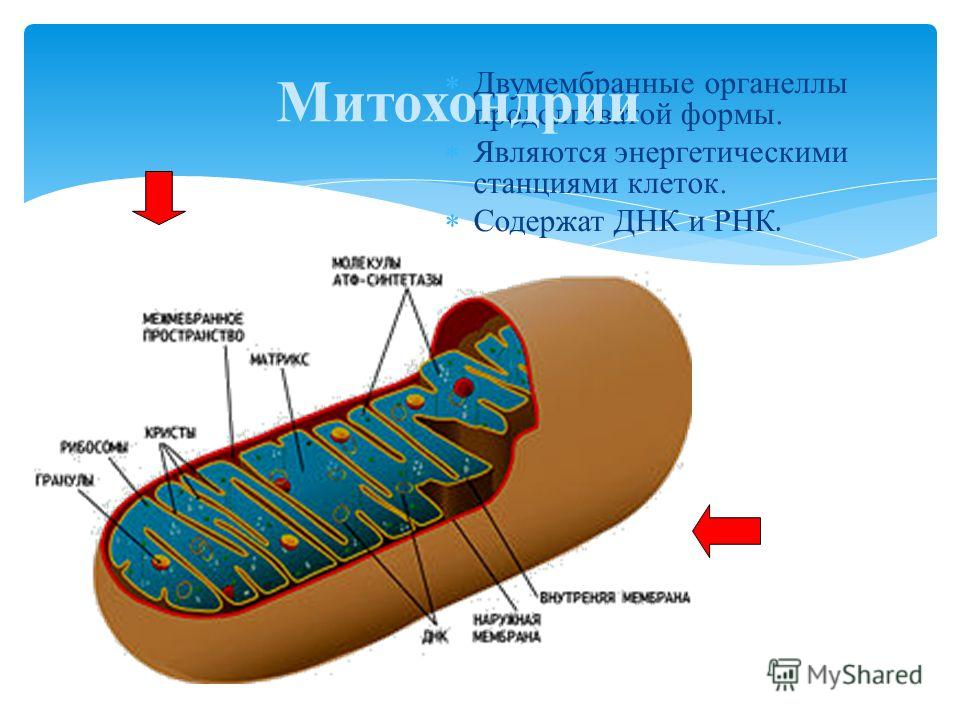 Чтобы подтвердить, деградации, более трудоемкий Вестерн-блоттинга должны быть завершены к β-галактозидазы 13, и это позволяет деградации количественное белка в небольших популяциях, подсчитанных червей. Блот плотности западные группы может быть определена количественно с помощью ImageJ 46. Другим ограничением этого метода является то, что использование трансгенных белков не сообщает, как в физиологических мишеней деградации и для таких ответов протеомические и / или целевые гипотеза приводом Вестерн-блоты должны быть использованы 13. И, наконец, как синтез белка в трансгенных используемого в этих исследованиях выключается в юношеском возрасте 19р>, другие методы должны быть использованы, когда желающих изучить изменения в синтезе белков мышечной в полностью развитой C. Элеганс мышцы; например, стабильные или радиоактивные изотопные методы.
Чтобы подтвердить, деградации, более трудоемкий Вестерн-блоттинга должны быть завершены к β-галактозидазы 13, и это позволяет деградации количественное белка в небольших популяциях, подсчитанных червей. Блот плотности западные группы может быть определена количественно с помощью ImageJ 46. Другим ограничением этого метода является то, что использование трансгенных белков не сообщает, как в физиологических мишеней деградации и для таких ответов протеомические и / или целевые гипотеза приводом Вестерн-блоты должны быть использованы 13. И, наконец, как синтез белка в трансгенных используемого в этих исследованиях выключается в юношеском возрасте 19р>, другие методы должны быть использованы, когда желающих изучить изменения в синтезе белков мышечной в полностью развитой C. Элеганс мышцы; например, стабильные или радиоактивные изотопные методы.
вовлечение митохондриальной дыхательной цепи
Biochem J. 2005 1 марта; 386 (Pt 2): 255–261.
Масато Кацуяма, отдел фармакологии
Медицинский университет префектуры, Киото 602-8566, Япония
ChunYuan Fan
* Кафедра фармакологии Медицинского университета префектуры Киото, Киото 602-8566, Япония
Нориаки Аракава
* Кафедра фармакологии Медицинского университета префектуры Киото , Киото 602-8566, Япония
Тору Нисинака
* Кафедра фармакологии Медицинского университета префектуры Киото, Киото 602-8566, Япония
Макото Миягиши
† Кафедра химии и биотехнологии инженерного факультета Университета Токио, Токио 113-8656, Япония
‡ Исследовательский центр функции генов, Национальный институт передовых промышленных наук и технологий, Научный городок Цукуба 305-8562, J apan
Кадзунари Тайра
† Кафедра химии и биотехнологии Инженерной школы Токийского университета, Токио 113-8656, Япония
‡ Исследовательский центр функции генов, Национальный институт передовых промышленных наук и технологий, Научный город Цукуба 305 -8562, Япония
Тихиро Ябе-Нишимура
* Кафедра фармакологии Медицинского университета префектуры Киото, Киото 602-8566, Япония
* Кафедра фармакологии Медицинского университета префектуры Киото, Киото 602-8566, Япония
† Департамент химии и биотехнологии, Школа инженерии, Токийский университет, Токио 113-8656, Япония
‡ Центр исследования функции генов, Национальный институт передовых промышленных наук и технологий, Научный городок Цукуба 305-8562, Япония
Получено 2004 12 июля; Пересмотрено 14 сентября 2004 г . ; Принята в печать 18 октября 2004 г.
Abstract
НАДФН-оксидаза является основным источником образования супероксида в сердечно-сосудистых тканях. Мы и другие сообщили, что PG (простагландин) F 2α , PDGF (фактор роста тромбоцитов) и ангиотензин II вызывают гипертрофию гладкомышечных клеток сосудов за счет индукции NOX1 (НАДФН-оксидаза 1), каталитической субъединицы НАДФН-оксидазы. Мы обнаружили, что DPI (дифенилен иодоний), ингибитор флавопротеинов, включая саму NADPH-оксидазу, почти полностью подавлял индукцию мРНК NOX1 PGF 2α или PDGF в линии гладкомышечных клеток сосудов крысы, A7r5.Исследование места действия DPI с использованием различных ингибиторов позволило предположить участие митохондриального окислительного фосфорилирования в PGF 2α — или индуцированное PDGF увеличение мРНК NOX1. В анализе репортерной люциферазы активация CRE (cAMP-response element) -зависимая транскрипция гена с помощью PGF 2α ослаблялась олигомицином, ингибитором митохондриальной F или F 1 -АТФазы.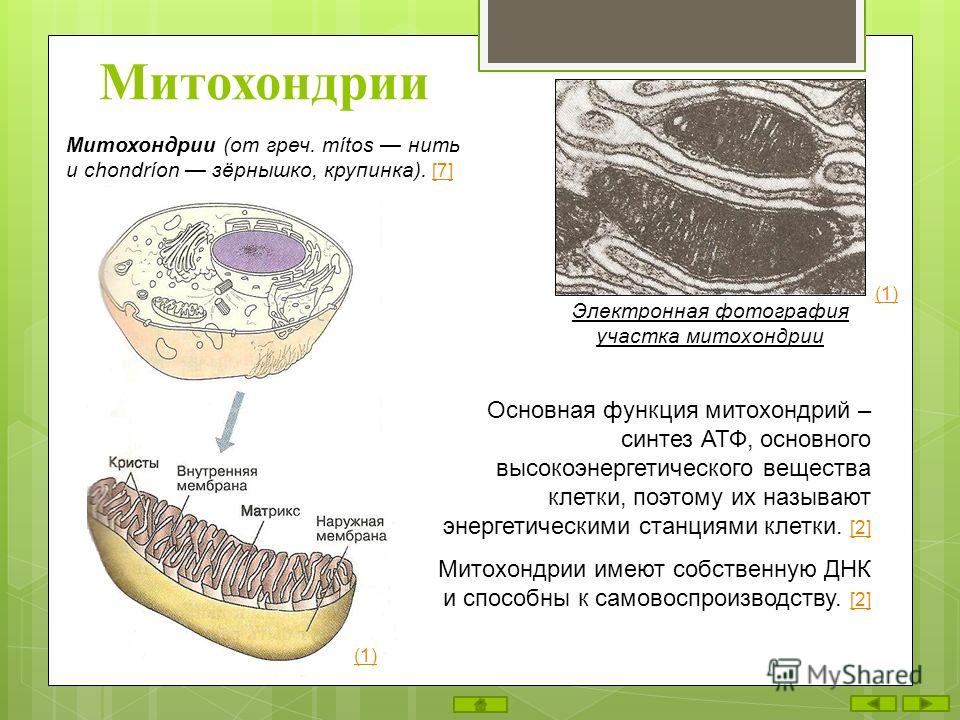 Олигомицин и другие митохондриальные ингибиторы также подавляли индуцированное PGF 2α фосфорилирование ATF (активирующий фактор транскрипции) -1, фактора транскрипции семейства CREB (CRE-связывающий белок) / ATF.Подавление гена ATF-1 за счет РНК-интерференции значительно снижает индукцию NOX1 PGF 2α или PDGF, тогда как сверхэкспрессия ATF-1 восстанавливает индукцию NOX1, подавляемую олигомицином. Взятые вместе, ATF-1 может играть ключевую роль в повышении регуляции NOX1 в гладкомышечных клетках сосудов крыс.
Олигомицин и другие митохондриальные ингибиторы также подавляли индуцированное PGF 2α фосфорилирование ATF (активирующий фактор транскрипции) -1, фактора транскрипции семейства CREB (CRE-связывающий белок) / ATF.Подавление гена ATF-1 за счет РНК-интерференции значительно снижает индукцию NOX1 PGF 2α или PDGF, тогда как сверхэкспрессия ATF-1 восстанавливает индукцию NOX1, подавляемую олигомицином. Взятые вместе, ATF-1 может играть ключевую роль в повышении регуляции NOX1 в гладкомышечных клетках сосудов крыс.
Ключевые слова: активирующий фактор транскрипции 1 (ATF-1), митохондрия, НАДФН-оксидаза, NOX1, простагландин F 2α
Сокращения: AP1, активирующий белок 1; ATF, активирующий фактор транскрипции; CRE, элемент cAMP-ответа; CREB, CRE-связывающий белок; DMEM, среда Игла, модифицированная Дульбекко; DPI, дифенилен иодоний; ds, двухцепочечный; ERK, киназа, регулируемая внеклеточными сигналами; FBS, фетальная бычья сыворотка; MnTBAP, Mn (III) тетракис (4-бензойная кислота) порфирин хлорид; МТТ, 3- (4,5-диметилтиазол-2-ил) -2,5-дифенил-2 H -тетразолий бромид; NOS, синтаза оксида азота; NOX1, НАДФН-оксидаза 1; PDGF, фактор роста тромбоцитов; PG, простагландин; PKCε, протеинкиназа Cε; СОД, супероксиддисмутаза; VSMC, гладкомышечные клетки сосудов
ВВЕДЕНИЕ
ROS (активные формы кислорода), включая супероксид (O 2 — ) и пероксид водорода (H 2 O 2 ), признаны важными сигнальными молекулами в сердечно-сосудистые ткани.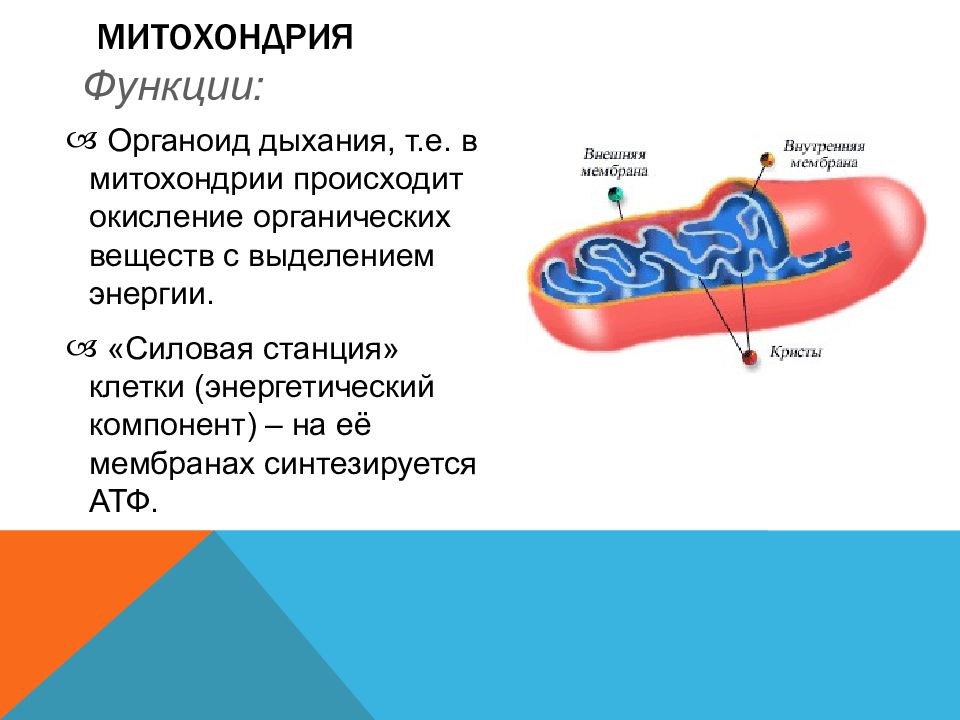 Было показано, что НАДФН-оксидазы являются основным источником O 2 – в сосудистых клетках и миоцитах [1–4]. Гомологи каталитической субъединицы НАДФН-оксидазы фагоцитов (gp91 phox ; NOX2) были обнаружены в VSMC (гладкомышечные клетки сосудов). Среди них NOX1 (НАДФН-оксидаза 1) участвует в патогенезе артериосклероза и реперфузионного повреждения, поскольку опосредует пролиферацию и гипертрофию VSMC [5–7]. Экспрессия NOX1 увеличивается при стимуляции PDGF (тромбоцитарным фактором роста), ангиотензином II, сложным эфиром форбола или FBS (фетальной бычьей сывороткой) [5,6].Ранее мы сообщали, что PG (простагландин) F 2α , один из первичных простаноидов, генерируемых в сосудистой ткани, вызывает гипертрофию VSMC за счет индукции NOX1 и последующего увеличения поколения O 2 — [7]. Таким образом, оказывается, что O 2 —, полученный из НАДФН-оксидазы, служит сигнальной молекулой, которая вызывает гипертрофию сосудов.
Было показано, что НАДФН-оксидазы являются основным источником O 2 – в сосудистых клетках и миоцитах [1–4]. Гомологи каталитической субъединицы НАДФН-оксидазы фагоцитов (gp91 phox ; NOX2) были обнаружены в VSMC (гладкомышечные клетки сосудов). Среди них NOX1 (НАДФН-оксидаза 1) участвует в патогенезе артериосклероза и реперфузионного повреждения, поскольку опосредует пролиферацию и гипертрофию VSMC [5–7]. Экспрессия NOX1 увеличивается при стимуляции PDGF (тромбоцитарным фактором роста), ангиотензином II, сложным эфиром форбола или FBS (фетальной бычьей сывороткой) [5,6].Ранее мы сообщали, что PG (простагландин) F 2α , один из первичных простаноидов, генерируемых в сосудистой ткани, вызывает гипертрофию VSMC за счет индукции NOX1 и последующего увеличения поколения O 2 — [7]. Таким образом, оказывается, что O 2 —, полученный из НАДФН-оксидазы, служит сигнальной молекулой, которая вызывает гипертрофию сосудов.
С другой стороны, имеется мало информации о точных молекулярных механизмах, лежащих в основе повышающей регуляции экспрессии гена NOX1 в VSMC.Мы обнаружили, что ингибитор флавопротеинов DPI (дифенилен иодоний) заметно подавляет индукцию мРНК NOX1 PGF 2α , PDGF, FBS или PMA. Поскольку DPI обычно используется в качестве ингибитора НАДФН-оксидазы путем образования аддуктов с восстановленным флавином [8–10], были проведены эксперименты для выяснения места действия DPI на сигнальном пути, ведущем к индукции экспрессии гена NOX1. В данной статье мы сообщаем о первых доказательствах роли митохондрий и ATF (активирующий фактор транскрипции) -1, члена семейства CREB (белок, связывающий цАМФ-элемент) / ATF, в регуляции NOX1 крыс. экспрессия гена.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
[α- 32 P] UTP и [α- 32 P] dCTP (3000 Ки / ммоль) были получены от ICN Biomedicals (Коста-Меса, Калифорния, США). PDGF-AB, PMA, хлорид DPI, тирон, ротенон, антимицин A и олигомицин были приобретены у Sigma (Сент-Луис, Миссури, США).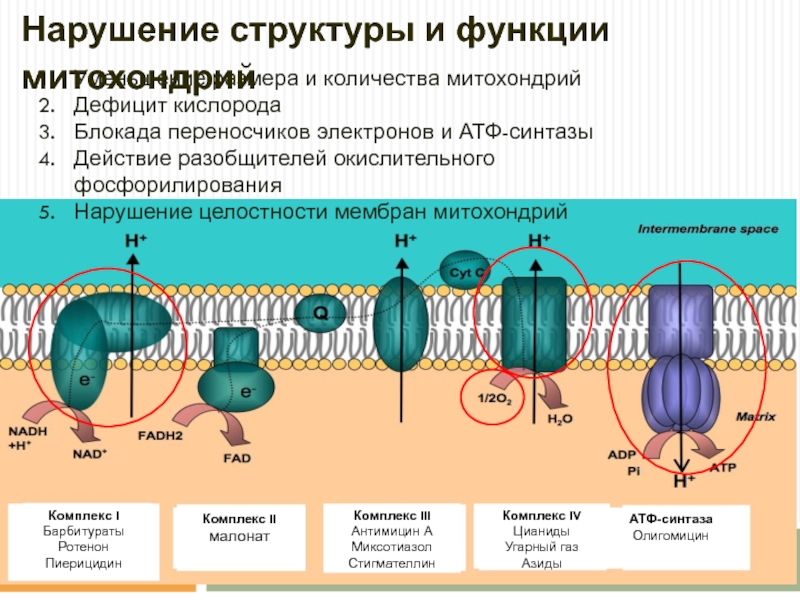 MnTBAP [Mn (III) тетракис (4-бензойная кислота) хлорид порфирина], EUK-8 и CCCP (карбонилцианид m -хлорфенилгидразон) были получены от Calbiochem-Novabiochem (Сан-Диего, Калифорния, США.С.А.). Комбинации PGF 2α , дисульфата G418 и ингибиторов протеазы были приобретены в Nacalai Tesque (Киото, Япония). Плазмиды, содержащие слитую ДНК специфического энхансерного элемента и люциферазы (системы профилирования пути ртути), были получены от Clontech (Пало-Альто, Калифорния, США).
MnTBAP [Mn (III) тетракис (4-бензойная кислота) хлорид порфирина], EUK-8 и CCCP (карбонилцианид m -хлорфенилгидразон) были получены от Calbiochem-Novabiochem (Сан-Диего, Калифорния, США.С.А.). Комбинации PGF 2α , дисульфата G418 и ингибиторов протеазы были приобретены в Nacalai Tesque (Киото, Япония). Плазмиды, содержащие слитую ДНК специфического энхансерного элемента и люциферазы (системы профилирования пути ртути), были получены от Clontech (Пало-Альто, Калифорния, США).
Нозерн-блоттинг
Клетки A7r5, полученные от A.T.C.C. (Манассас, Вирджиния, США) поддерживали в DMEM (среда Игла, модифицированная Дульбекко) с добавлением 10% FBS. Перед добавлением реагентов клетки культивировали в среде DMEM, содержащей 0.5% FBS в течение 48 часов. Затем клетки инкубировали со 100 нМ PGF 2α , 20 нг / мл PDGF-AB, 10% FBS или 100 нМ PMA в присутствии или в отсутствие различных ингибиторов в течение 24 часов. Выделение РНК и Нозерн-блоттинг выполняли в основном, как описано в [7].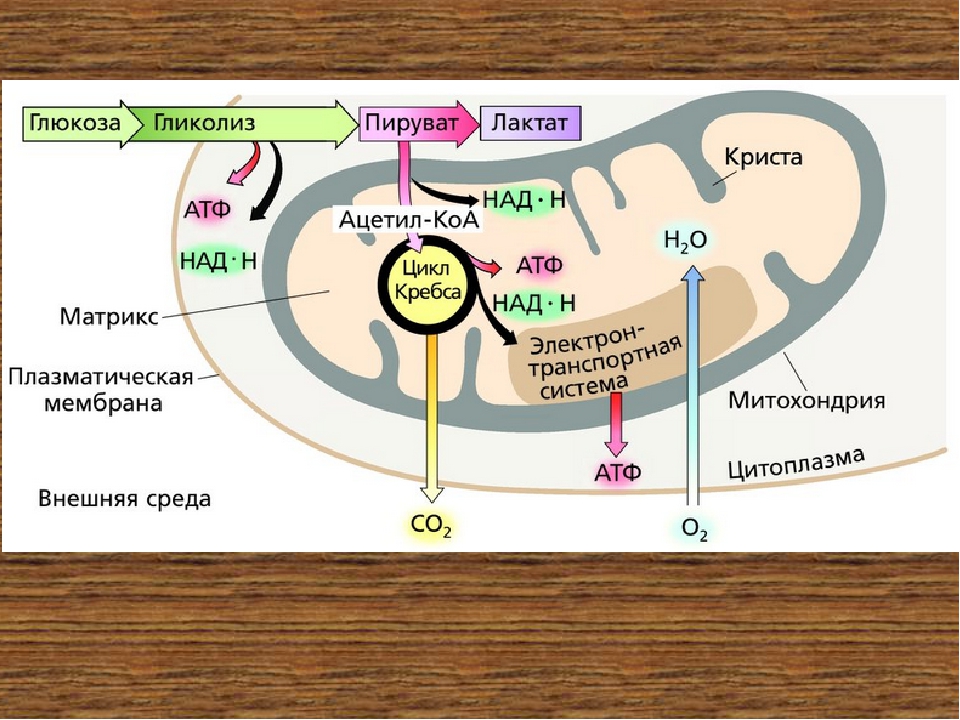 Жизнеспособность клеток определяли с использованием МТТ [3- (4,5-диметилтиазол-2-ил) -2,5-дифенил-2 H -тетразолийбромида], по существу, как описано ранее [11].
Жизнеспособность клеток определяли с использованием МТТ [3- (4,5-диметилтиазол-2-ил) -2,5-дифенил-2 H -тетразолийбромида], по существу, как описано ранее [11].
Люциферазный анализ
Клетки A7r5 высевали в шестилуночные планшеты и культивировали в течение 24 часов в среде DMEM с добавлением 10% FBS.Люциферазные плазмиды (1 мкг / лунка) и контрольный вектор pSV-β-галактозидазы (0,5 мкг / лунка; Promega, Мэдисон, Висконсин, США) котрансфицировали в клетки с помощью реагента для трансфекции FuGENE 6 (Roche, Индианаполис, Индиана, США). ). После трансфекции клетки культивировали в течение 24 часов в среде DMEM с добавлением 10% FBS, а затем в отсутствие FBS в течение еще 24 часов. Затем клетки инкубировали со 100 нМ PGF 2α в присутствии или в отсутствие 2 мкг / мл олигомицина в течение 24 часов. Готовили клеточные лизаты и определяли активность люциферазы, нормированную на активность β-галактозидазы, в соответствии с инструкциями производителя.
Вестерн-блоттинг
Клетки A7r5, культивированные в отсутствие FBS в течение 48 часов, предварительно обрабатывали митохондриальными ингибиторами в течение 30 минут, а затем инкубировали со 100 нМ PGF 2α в течение 5 минут.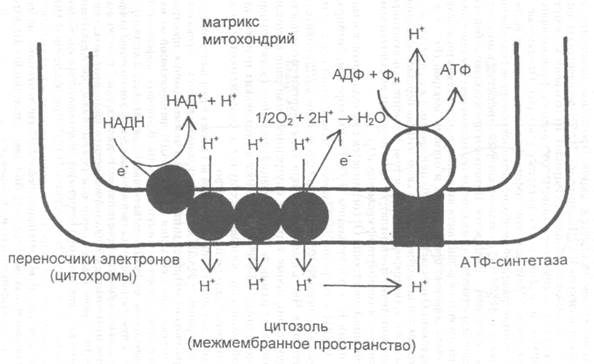 Ядерные экстракты готовили, как описано ранее [11,12]. Вкратце, клетки суспендировали в буфере A (10 мМ Hepes / KOH, pH 7,9, 1,5 мМ MgCl 2 и 10 мМ KCl), содержащем коктейли ингибиторов протеаз, 0,5 мМ дитиотреитол, 1 мМ NaF, 20 мМ β-глицерофосфат и 1 мМ На 3 ВО 4 .После центрифугирования осадки ресуспендировали в буфере C (20 мМ Hepes / KOH, pH 7,9, 25% глицерин, 420 мМ NaCl, 1,5 мМ MgCl 2 и 0,2 мМ EDTA), содержащем описанные выше ингибиторы. Супернатанты, полученные после центрифугирования, использовали в качестве ядерных экстрактов. Экстракты (10 мкг) разделяли с помощью SDS / 12,5% PAGE и переносили на мембраны из PVDF (Millipore, Bedford, MA, USA). Мембраны инкубировали с антителом против фосфо-CREB (реактивным с фосфорилированными формами CREB и ATF-1; Cell Signaling Technology, Beverly, MA, U.S.A.) или моноклональное антитело против ATF-1 (25C10G; Santa Cruz Biotechnology, Санта-Крус, Калифорния, США), а затем с антикроличьим IgG или антимышиным IgG, конъюгированным с пероксидазой хрена соответственно.
Ядерные экстракты готовили, как описано ранее [11,12]. Вкратце, клетки суспендировали в буфере A (10 мМ Hepes / KOH, pH 7,9, 1,5 мМ MgCl 2 и 10 мМ KCl), содержащем коктейли ингибиторов протеаз, 0,5 мМ дитиотреитол, 1 мМ NaF, 20 мМ β-глицерофосфат и 1 мМ На 3 ВО 4 .После центрифугирования осадки ресуспендировали в буфере C (20 мМ Hepes / KOH, pH 7,9, 25% глицерин, 420 мМ NaCl, 1,5 мМ MgCl 2 и 0,2 мМ EDTA), содержащем описанные выше ингибиторы. Супернатанты, полученные после центрифугирования, использовали в качестве ядерных экстрактов. Экстракты (10 мкг) разделяли с помощью SDS / 12,5% PAGE и переносили на мембраны из PVDF (Millipore, Bedford, MA, USA). Мембраны инкубировали с антителом против фосфо-CREB (реактивным с фосфорилированными формами CREB и ATF-1; Cell Signaling Technology, Beverly, MA, U.S.A.) или моноклональное антитело против ATF-1 (25C10G; Santa Cruz Biotechnology, Санта-Крус, Калифорния, США), а затем с антикроличьим IgG или антимышиным IgG, конъюгированным с пероксидазой хрена соответственно.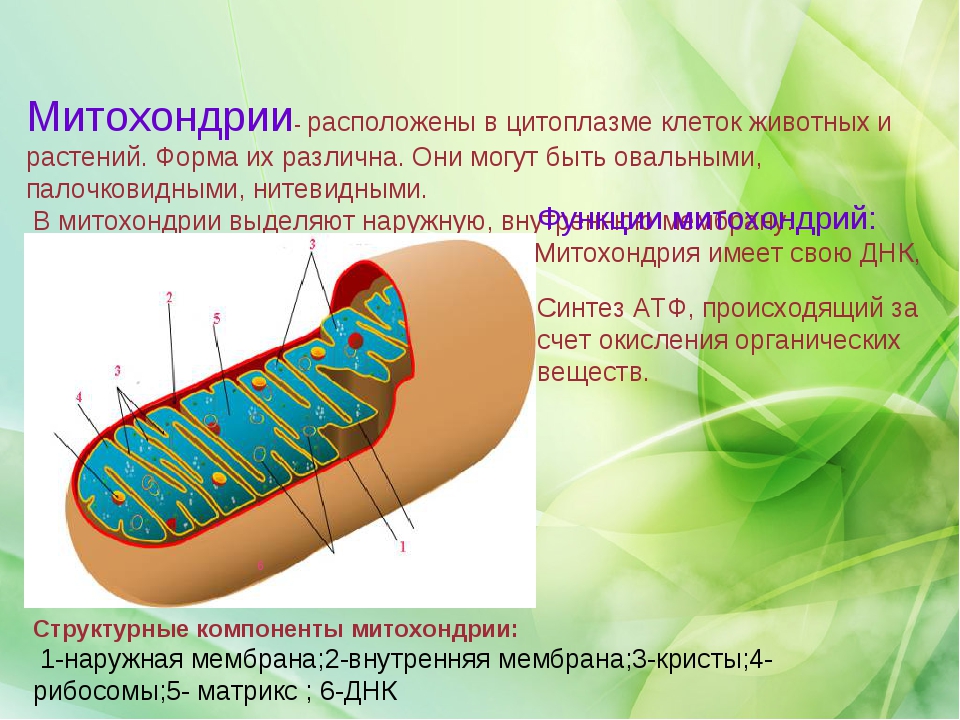 Иммунореактивные полосы детектировали с помощью системы ECL® (усиленная хемилюминесценция) Plus (Amersham Biosciences, Пискатауэй, Нью-Джерси, США).
Иммунореактивные полосы детектировали с помощью системы ECL® (усиленная хемилюминесценция) Plus (Amersham Biosciences, Пискатауэй, Нью-Джерси, США).
Синтез ds (двухцепочечной) РНК против ATF-1
Вектор экспрессии dsRNA, pcPURU6icassette, был сконструирован путем вставки гена пуромицин N -ацетилтрансферазы в вектор pU6icassette [13,14], который содержит промотор U6 человека и два сайта Bfu AI ( Bsp MI).Была разработана дцРНК против ATF-1, которая нацелена на нуклеотиды 247–267 клона кДНК крысы {«type»: «entrez-нуклеотид», «attrs»: {«text»: «BU671705», «term_id»: «23400599 «,» term_text «:» BU671705 «}} BU671705. Смысловые или антисмысловые олигонуклеотиды, содержащие последовательность шпильки, последовательность терминатора и выступающие последовательности, были синтезированы, амплифицированы с помощью ПЦР, расщеплены Bfu AI и вставлены в сайт Bfu AI кассеты pcPURU6ic.
Создание клонов, стабильно экспрессирующих дцРНК против ATF-1
Вектор экспрессии дцРНК (pcPURU6icassette, содержащий последовательность дцРНК против ATF-1) трансфицировали в клетки A7r5, как описано выше. Стабильные трансфектанты отбирали клонированием одной клетки в присутствии пуромицина (5 мкг / мл). Для имитации трансфекции вектор pcPURU6icassette трансфицировали и отбирали пуромицином.
Стабильные трансфектанты отбирали клонированием одной клетки в присутствии пуромицина (5 мкг / мл). Для имитации трансфекции вектор pcPURU6icassette трансфицировали и отбирали пуромицином.
Создание клонов, стабильно экспрессирующих ATF-1
Кодирующую область кДНК ATF-1 крысы амплифицировали с помощью RT (обратной транскриптазы) -PCR и вставляли в pcDNA3. Плазмиду трансфецировали в клетки A7r5, как описано выше, и стабильные трансфектанты отбирали путем клонирования одной клетки в присутствии G418 (1 мг / мл).
Статистический анализ
Значения выражены как средние значения ± стандартная ошибка среднего. Статистический анализ проводился с помощью теста Стьюдента t . Для нескольких групп лечения применяли однофакторный дисперсионный анализ с последующим тестом Бонферрони t .
РЕЗУЛЬТАТЫ
DPI подавляет индукцию мРНК NOX1
Как мы сообщали ранее, PGF 2α увеличивает уровни мРНК NOX1 в VSMCs крыс, A7r5 [7].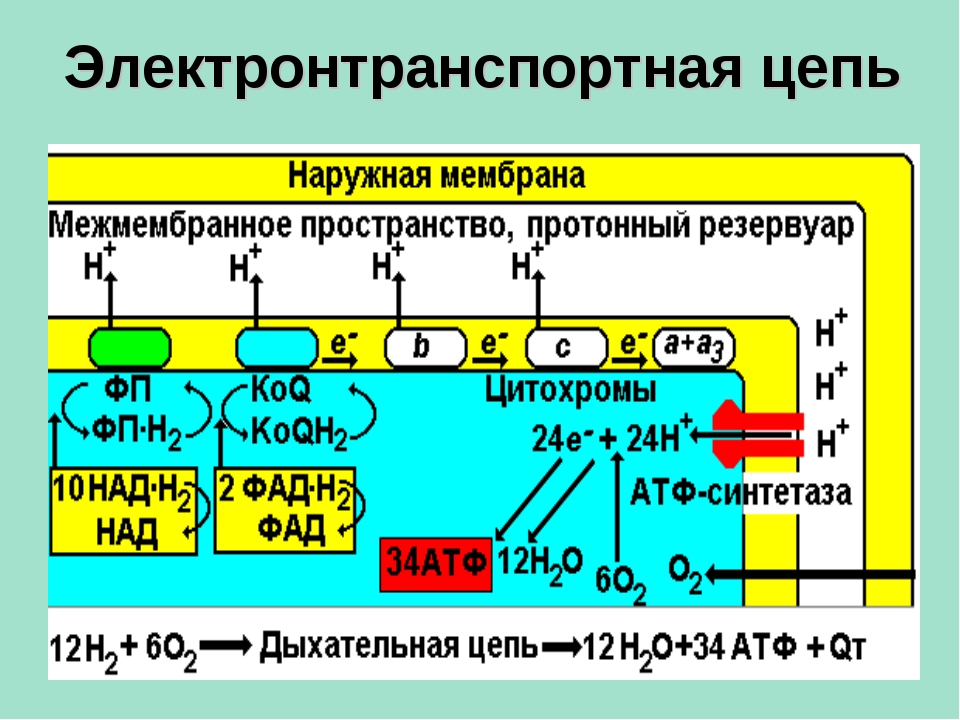 В ходе исследования сигнальных путей, которые опосредуют индуцированную PGF 2α экспрессию NOX1, мы обнаружили, что 100 нМ DPI, ингибитор НАДФН-оксидазы, почти полностью подавлял индукцию мРНК NOX1 с помощью PGF 2α .DPI также подавлял увеличение мРНК NOX1, индуцированное PDGF, 10% FBS или PMA (A). Анализ МТТ показал, что более 85% клеток были жизнеспособными, когда клетки инкубировали в присутствии 100 нМ DPI в течение 24 часов (результаты не показаны). В этих клетках четко наблюдалась индукция c-fos 10% FBS (B). Эти данные свидетельствуют о том, что подавляющий эффект DPI на индукцию NOX1 не связан с повреждением клеток.
В ходе исследования сигнальных путей, которые опосредуют индуцированную PGF 2α экспрессию NOX1, мы обнаружили, что 100 нМ DPI, ингибитор НАДФН-оксидазы, почти полностью подавлял индукцию мРНК NOX1 с помощью PGF 2α .DPI также подавлял увеличение мРНК NOX1, индуцированное PDGF, 10% FBS или PMA (A). Анализ МТТ показал, что более 85% клеток были жизнеспособными, когда клетки инкубировали в присутствии 100 нМ DPI в течение 24 часов (результаты не показаны). В этих клетках четко наблюдалась индукция c-fos 10% FBS (B). Эти данные свидетельствуют о том, что подавляющий эффект DPI на индукцию NOX1 не связан с повреждением клеток.
( A ) Влияние DPI на индукцию мРНК NOX1.Клетки A7r5, поддерживаемые в DMEM с 0,5% FBS в течение 48 ч, инкубировали со 100 нМ PGF 2α , 20 нг / мл PDGF-AB, 10% FBS или 100 нМ PMA в течение 24 ч в присутствии или отсутствии 100 нМ. DPI. Демонстрируется репрезентативный авторадиограф трех экспериментов.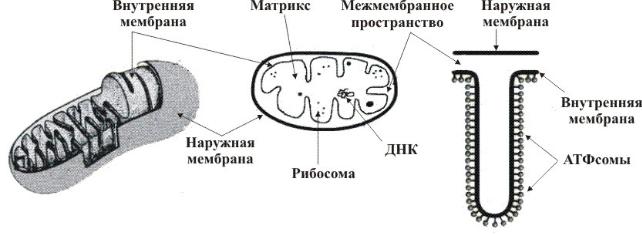 Под блотом показаны средние уровни экспрессии NOX1, нормализованные к 28 S РНК. ( B ) Влияние DPI на индукцию мРНК c-fos с помощью FBS. Клетки A7r5 с остановкой роста инкубировали с 100 нМ DPI в течение указанного времени, а затем стимулировали 10% FBS в течение 30 минут.Нозерн-блоттинг выполняли, как описано в экспериментальном разделе.
Под блотом показаны средние уровни экспрессии NOX1, нормализованные к 28 S РНК. ( B ) Влияние DPI на индукцию мРНК c-fos с помощью FBS. Клетки A7r5 с остановкой роста инкубировали с 100 нМ DPI в течение указанного времени, а затем стимулировали 10% FBS в течение 30 минут.Нозерн-блоттинг выполняли, как описано в экспериментальном разделе.
Поглотители O
2 — не влияют на индукцию мРНК NOX1 Для дальнейшего выяснения влияния DPI на индукцию NOX1 мы сначала исследовали, являются ли поглотители O 2 —, продукты реакции НАДФН-оксидаза может влиять на экспрессию гена NOX1. MnTBAP, проникающий в клетки миметик SOD (супероксиддисмутаза) и акцептор пероксинитрита, и тайрон, проникающий в клетки акцептор O 2 —, не влияли на индукцию NOX1 PGF 2α .Более того, EUK-8, синтетический сален-марганцевый комплекс с высокой активностью по улавливанию SOD, каталазы и оксирадикала, не показал эффекта на индукцию NOX1 с помощью PGF 2α ().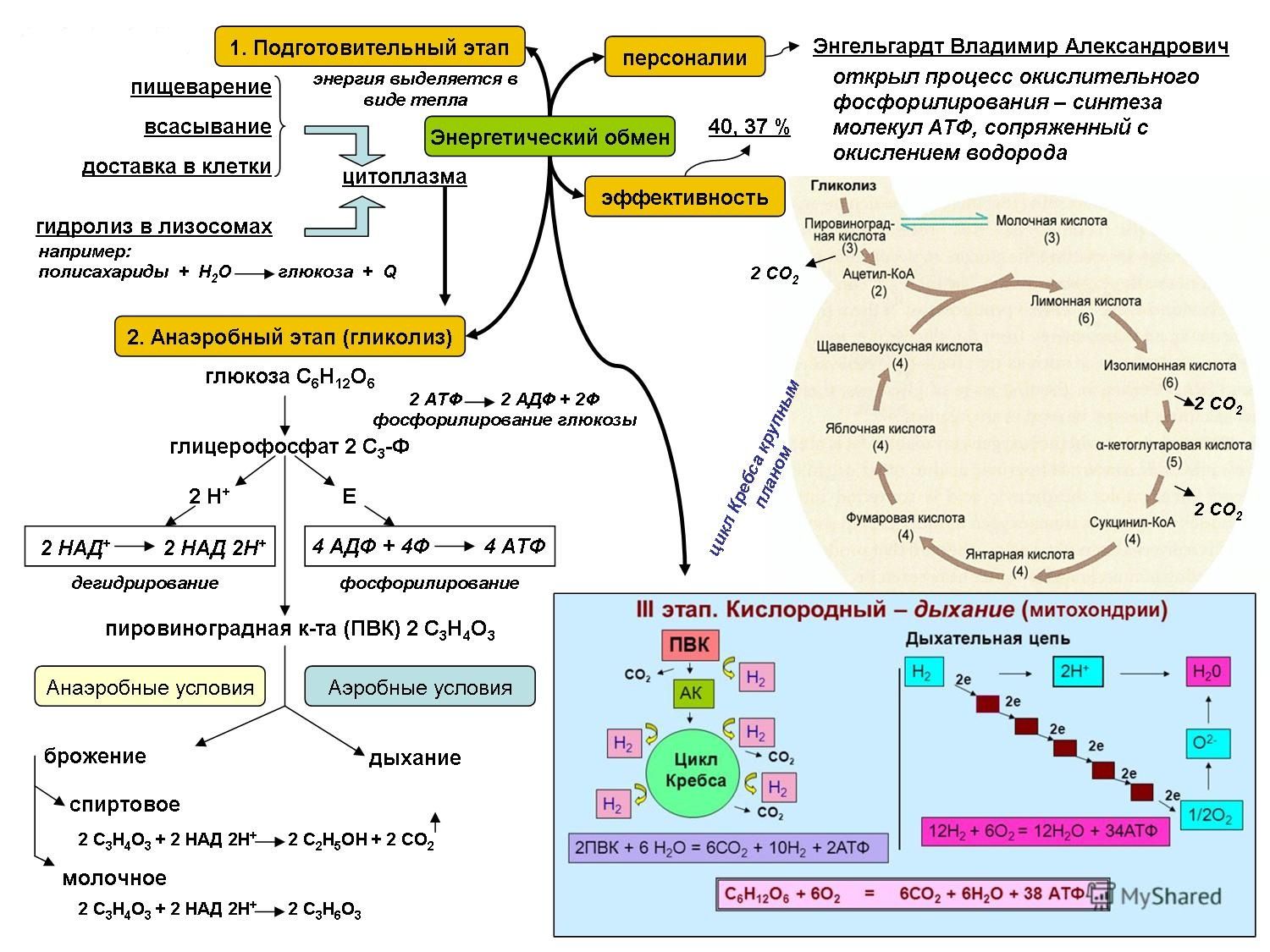 Эти результаты предполагают, что индукция NOX1 не опосредуется O 2 —, H 2 O 2 или оксирадикалами, и что влияние DPI на индукцию NOX1 не связано с ингибированием активности NADPH-оксидазы с помощью DPI. . DPI также известен как ингибитор NOS (синтазы оксида азота) [15]. N G -монометил-L-аргинин (L-NMMA), ингибитор NOS, однако, не подавлял индукцию NOX1 PGF 2α (результаты не показаны). Таким образом было исключено участие NO в индукции мРНК NOX1.
Эти результаты предполагают, что индукция NOX1 не опосредуется O 2 —, H 2 O 2 или оксирадикалами, и что влияние DPI на индукцию NOX1 не связано с ингибированием активности NADPH-оксидазы с помощью DPI. . DPI также известен как ингибитор NOS (синтазы оксида азота) [15]. N G -монометил-L-аргинин (L-NMMA), ингибитор NOS, однако, не подавлял индукцию NOX1 PGF 2α (результаты не показаны). Таким образом было исключено участие NO в индукции мРНК NOX1.
Клетки A7r5, поддерживаемые в DMEM с 0,5% FBS в течение 48 ч, инкубировали со 100 мкМ MnTBAP, 10 мМ тироном. или 50 мкМ EUK-8 в течение 24 ч в присутствии 100 нМ PGF 2α .Демонстрируется репрезентативный авторадиограф трех экспериментов. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотом.
Ингибиторы митохондриальной дыхательной цепи подавляют индукцию мРНК NOX1.
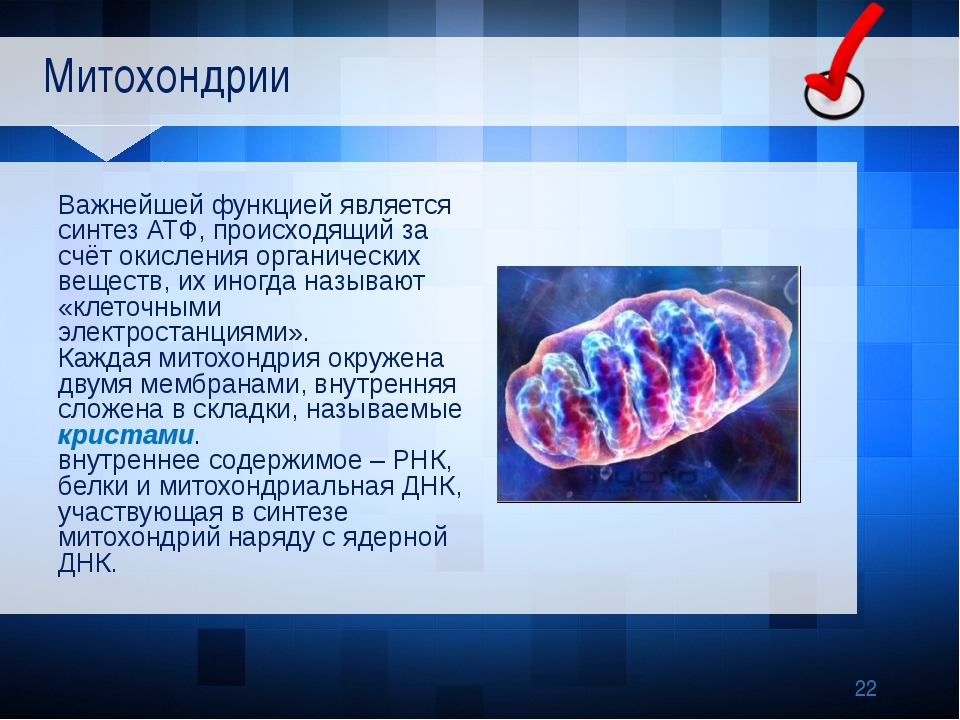
DPI ингибирует комплекс I в дыхательной цепи митохондрий в дополнение к НАДФН-оксидазе [16]. Поэтому далее было исследовано участие системы транспорта электронов в индукции NOX1. Ротенон и антимицин А, ингибиторы комплексов I и III соответственно, почти полностью блокировали индукцию NOX1 PGF 2α .Точно так же индукция NOX1 с помощью PGF 2α подавлялась ингибитором F o F 1 -АТФазы, олигомицином и разобщителем окислительного фосфорилирования CCCP (A). Все эти ингибиторы также подавляли индуцированную PDGF экспрессию NOX1 (B). В присутствии этих митохондриальных ингибиторов индукция c-fos с помощью 10% FBS сохранялась (см. Дополнительный рисунок 1 на http://www.BiochemJ.org/bj/386/bj3860255add.htm). В ходе проточно-цитометрического анализа с использованием флуоресцентного красителя JC-1 мы подтвердили, что такая же концентрация CCCP заметно снижает потенциал митохондриальной мембраны (результаты не показаны).В анализе МТТ более 75% клеток были жизнеспособными в присутствии ротенона или CCCP. Что касается антимицина А и олигомицина, эти ингибиторы не влияли на жизнеспособность клеток (результаты не показаны). Таким образом, эти результаты предполагают, что ингибирование митохондриальной дыхательной цепи подавляет индукцию NOX1 с помощью PGF 2α и PDGF.
Что касается антимицина А и олигомицина, эти ингибиторы не влияли на жизнеспособность клеток (результаты не показаны). Таким образом, эти результаты предполагают, что ингибирование митохондриальной дыхательной цепи подавляет индукцию NOX1 с помощью PGF 2α и PDGF.
Клетки A7r5, поддерживаемые в DMEM с 0,5% FBS в течение 48 ч, инкубировали с 500 нМ ротеноном, 100 нг / мл антимицина A, 2 мкг / мл олигомицина или 10 мкг / мл олигомицина. мкМ CCCP в течение 24 ч в присутствии 100 нМ PGF 2α ( A ) или 20 нг / мл PDGF-AB ( B ).Демонстрируются репрезентативные авторадиограммы трех экспериментов. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотами.
CRE (cAMP-response element) -зависимая активация транскрипции с помощью PGF
2α подавляется олигомицином Чтобы прояснить путь передачи сигнала, который опосредует митохондрии в ядро, мы проверили факторы транскрипции, которые активируются PGF 2α и ингибируется митохондриальными ингибиторами. Клетки A7r5 трансфицировали плазмидами, содержащими специфические энхансерные элементы, слитые с ДНК люциферазы, и стимулировали PGF 2α в присутствии или в отсутствие олигомицина. Среди сайта связывания AP1 (активирующего протеина 1), CRE, элемента ответа на глюкокортикоиды, элемента теплового шока, сайта связывания NF-κB (ядерного фактора κB) и элемента ответа сыворотки только CRE-зависимая транскрипция усиливалась PGF . 2α более чем в 2 раза по сравнению с контролем. Как показано на фиг.1, AP1-зависимая транскрипционная активность была немного усилена PGF 2α , который, по-видимому, был связан с сопутствующей индукцией c-fos с помощью PGF 2α , который связывается с сайтами AP1 [17].Однако, в отличие от AP1-зависимой активации, CRE-зависимая транскрипционная активация PGF 2α значительно подавлялась олигомицином.
Клетки A7r5 трансфицировали плазмидами, содержащими специфические энхансерные элементы, слитые с ДНК люциферазы, и стимулировали PGF 2α в присутствии или в отсутствие олигомицина. Среди сайта связывания AP1 (активирующего протеина 1), CRE, элемента ответа на глюкокортикоиды, элемента теплового шока, сайта связывания NF-κB (ядерного фактора κB) и элемента ответа сыворотки только CRE-зависимая транскрипция усиливалась PGF . 2α более чем в 2 раза по сравнению с контролем. Как показано на фиг.1, AP1-зависимая транскрипционная активность была немного усилена PGF 2α , который, по-видимому, был связан с сопутствующей индукцией c-fos с помощью PGF 2α , который связывается с сайтами AP1 [17].Однако, в отличие от AP1-зависимой активации, CRE-зависимая транскрипционная активация PGF 2α значительно подавлялась олигомицином.
Люциферазные плазмиды, содержащие различные энхансерные элементы, котрансфицировали с помощью контрольного вектора β-галактозидазы в клетки A7r5. Активность люциферазы нормализовалась по активности β-галактозидазы. Результаты — это средние значения ± S.Э.М. ( n = 3). pTAL-Luc, контрольный вектор, лишенный энхансерного элемента. pAP1-Luc, люциферазная плазмида, содержащая сайт связывания AP1. pCRE-Luc, люциферазная плазмида, содержащая CRE. Открытые бары, контроль; закрытые бары, PGF 2α ; пунктирная полоса — олигомицин; заштрихованная полоса, PGF 2α + олигомицин. * P <0,001 по сравнению с контролем. † P <0,001 по сравнению с PGF 2α .
Активность люциферазы нормализовалась по активности β-галактозидазы. Результаты — это средние значения ± S.Э.М. ( n = 3). pTAL-Luc, контрольный вектор, лишенный энхансерного элемента. pAP1-Luc, люциферазная плазмида, содержащая сайт связывания AP1. pCRE-Luc, люциферазная плазмида, содержащая CRE. Открытые бары, контроль; закрытые бары, PGF 2α ; пунктирная полоса — олигомицин; заштрихованная полоса, PGF 2α + олигомицин. * P <0,001 по сравнению с контролем. † P <0,001 по сравнению с PGF 2α .
Ингибиторы митохондриальной дыхательной цепи подавляют фосфорилирование ATF-1 с помощью PGF
2α CRE-зависимая транскрипция опосредуется факторами транскрипции семейства CREB / ATF.Особый интерес представляет тот факт, что сообщалось об участии CREB и ATF-1 в индуцированной ангиотензином-II гипертрофии и индуцированном тромбином росте VSMC [18,19]. Поэтому мы исследовали, индуцирует ли PGF 2α фосфорилирование этих белков. Как показано на (A), белок 35 кДа был обнаружен в ядерных экстрактах клеток, стимулированных PGF 2α , с помощью антитела против фосфо-CREB, которое реагирует с фосфорилированными формами CREB и ATF-1. Хотя фосфорилированная форма CREB (43 кДа) не была обнаружена в этих клетках, зависимое от времени увеличение фосфорилирования ATF-1 наблюдалось до 15 мин.Эти данные предполагают, что PGF 2α вызывает фосфорилирование ATF-1, но не CREB. PDGF также вызывал фосфорилирование ATF-1 за аналогичный период времени (результаты не показаны). Затем были изучены эффекты митохондриальных ингибиторов на индуцированное PGF 2α фосфорилирование ATF-1. Как показано на (B), DPI и все другие ингибиторы митохондрий подавляли фосфорилирование ATF-1, индуцированное PGF 2α . DPI также подавлял индуцированное PDGF фосфорилирование ATF-1 (результаты не показаны).
Как показано на (A), белок 35 кДа был обнаружен в ядерных экстрактах клеток, стимулированных PGF 2α , с помощью антитела против фосфо-CREB, которое реагирует с фосфорилированными формами CREB и ATF-1. Хотя фосфорилированная форма CREB (43 кДа) не была обнаружена в этих клетках, зависимое от времени увеличение фосфорилирования ATF-1 наблюдалось до 15 мин.Эти данные предполагают, что PGF 2α вызывает фосфорилирование ATF-1, но не CREB. PDGF также вызывал фосфорилирование ATF-1 за аналогичный период времени (результаты не показаны). Затем были изучены эффекты митохондриальных ингибиторов на индуцированное PGF 2α фосфорилирование ATF-1. Как показано на (B), DPI и все другие ингибиторы митохондрий подавляли фосфорилирование ATF-1, индуцированное PGF 2α . DPI также подавлял индуцированное PDGF фосфорилирование ATF-1 (результаты не показаны).
( A ) Динамика фосфорилирования ATF-1 с помощью PGF 2α . Клетки A7r5 с недостатком сыворотки инкубировали со 100 нМ PGF 2α в течение указанного времени. ( B ) Эффекты различных ингибиторов дыхательной цепи митохондрий. Клетки A7r5, лишенные сыворотки, предварительно инкубировали с 500 нМ ротенона, 100 нг / мл антимицина A, 2 мкг / мл олигомицина, 10 мкМ CCCP или указанных концентраций DPI в течение 30 минут и стимулировали 100 нМ PGF 2α в течение 30 минут. 5 минут.Вестерн-блоттинг выполняли, как описано в экспериментальном разделе. Показаны репрезентативные результаты трех экспериментов. Усредненные относительные интенсивности полос для фосфорилированного ATF-1 показаны под блотами.
Клетки A7r5 с недостатком сыворотки инкубировали со 100 нМ PGF 2α в течение указанного времени. ( B ) Эффекты различных ингибиторов дыхательной цепи митохондрий. Клетки A7r5, лишенные сыворотки, предварительно инкубировали с 500 нМ ротенона, 100 нг / мл антимицина A, 2 мкг / мл олигомицина, 10 мкМ CCCP или указанных концентраций DPI в течение 30 минут и стимулировали 100 нМ PGF 2α в течение 30 минут. 5 минут.Вестерн-блоттинг выполняли, как описано в экспериментальном разделе. Показаны репрезентативные результаты трех экспериментов. Усредненные относительные интенсивности полос для фосфорилированного ATF-1 показаны под блотами.
Подавление гена ATF-1 ослабляет индукцию мРНК NOX1
Для дополнительной проверки участия ATF-1 в индукции экспрессии гена NOX1 в клетки A7r5 вводили дцРНК, нацеленную на последовательность мРНК ATF-1 крысы. После клонирования одной клетки трансфектантов были выделены два клона (RNAi-5 и RNAi-16), стабильно экспрессирующие дцРНК (A, левая панель).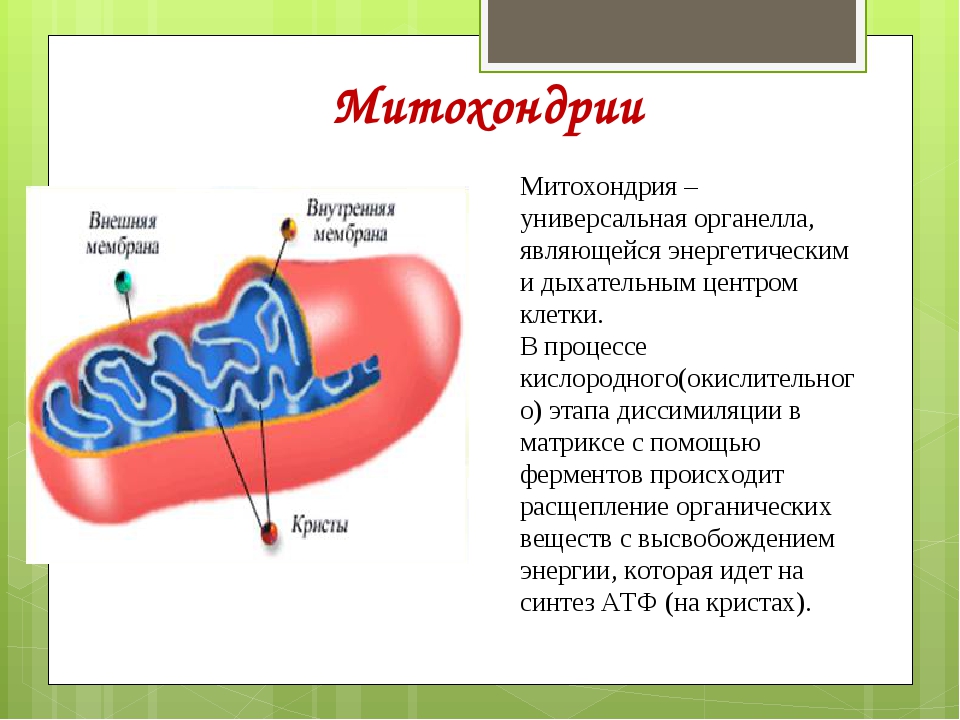 В RNAi-5 и RNAi-16 уровни ATF-1, но не CREB, были значительно снижены по сравнению с ложно трансфицированными клетками (A, правая панель). Как показано на (B), индукция мРНК NOX1 с помощью PGF 2α в этих клетках была почти полностью устранена. Кроме того, в этих клонах также ингибировалась индукция мРНК NOX1 с помощью PDGF, 10% FBS или PMA (C). В этих клонах четко наблюдалась индукция c-fos 10% FBS (см. Дополнительный рисунок 2 на http://www.BiochemJ.org/bj/386/bj3860255add.htm).
В RNAi-5 и RNAi-16 уровни ATF-1, но не CREB, были значительно снижены по сравнению с ложно трансфицированными клетками (A, правая панель). Как показано на (B), индукция мРНК NOX1 с помощью PGF 2α в этих клетках была почти полностью устранена. Кроме того, в этих клонах также ингибировалась индукция мРНК NOX1 с помощью PDGF, 10% FBS или PMA (C). В этих клонах четко наблюдалась индукция c-fos 10% FBS (см. Дополнительный рисунок 2 на http://www.BiochemJ.org/bj/386/bj3860255add.htm).
( A ) Экспрессия дцРНК против ATF-1 (левая панель) и подавление экспрессии ATF-1 (правая панель) в клонах РНКи-5 и РНКи-16. Тотальную РНК подвергали обратной транскрипции с помощью случайных нономеров, а фрагмент кДНК (57 п.н.) амплифицировали с помощью ПЦР. Продукт разделяли с помощью 15% PAGE. Вестерн-блоттинг выполняли, как описано в экспериментальном разделе. ( B ) Индукция мРНК NOX1 с помощью PGF 2α подавлялась в RNAi-5 и RNAi-16. ( C ) Индукция мРНК NOX1 с помощью PDGF, 10% FBS или PMA подавлялась в RNAi-5 и RNAi-16. Нозерн-блоттинг выполняли, как описано в экспериментальном разделе. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотами.
( C ) Индукция мРНК NOX1 с помощью PDGF, 10% FBS или PMA подавлялась в RNAi-5 и RNAi-16. Нозерн-блоттинг выполняли, как описано в экспериментальном разделе. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотами.
Сверхэкспрессия ATF-1 восстанавливает PGF
2α -индуцированную экспрессию NOX1, подавленную олигомициномДля изучения эффектов сверхэкспрессии ATF-1 на индукцию NOX1 была введена экспрессионная плазмида, содержащая кодирующую область кДНК ATF-1 крысы. в клетки A7r5.После клонирования одной клетки трансфектантов были выделены два клона, стабильно экспрессирующие ATF-1, ATF1-3 и ATF1-7 (A). Как показано на (B), индукция NOX1 PGF 2α в этих клонах наблюдалась в присутствии олигомицина. Эти находки, вместе с результатами подавления гена ATF-1, ясно показывают, что ATF-1 является важным фактором транскрипции, который опосредует экспрессию гена NOX1 в VSMC крыс.
Сверхэкспрессия ATF-1 восстанавливала PGF 2α -индуцированную экспрессию NOX1, подавленную олигомицином ( A ) Сверхэкспрессия ATF-1 в клонах ATF1-3 и ATF1-7. Вестерн-блоттинг выполняли, как описано в экспериментальном разделе. ( B ) На индукцию мРНК NOX1 с помощью PGF 2α не влиял олигомицин в ATF1-3 и ATF1-7. Нозерн-блоттинг выполняли, как описано в экспериментальном разделе. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотом.
Вестерн-блоттинг выполняли, как описано в экспериментальном разделе. ( B ) На индукцию мРНК NOX1 с помощью PGF 2α не влиял олигомицин в ATF1-3 и ATF1-7. Нозерн-блоттинг выполняли, как описано в экспериментальном разделе. Относительные уровни экспрессии NOX1, нормализованные к 28 S РНК, показаны под блотом.
ОБСУЖДЕНИЕ
Настоящие результаты предполагают, что ATF-1, фактор транскрипции семейства CREB / ATF, играет важную роль в индукции мРНК NOX1.Основные доказательства, представленные в настоящем исследовании, заключаются в следующем: (i) DPI, который ингибирует НАДФН-оксидазу в дополнение к комплексу I в дыхательной цепи митохондрий, почти полностью устраняет индукцию мРНК NOX1 PGF 2α , PDGF, FBS или PMA; (ii) увеличение мРНК NOX1, индуцированное PGF 2α или PDGF, ослаблялось ингибиторами дыхательной цепи митохондрий; (iii) CRE-зависимая активация транскрипции с помощью PGF 2α подавлялась олигомицином, ингибитором митохондриальной F o -АТФазы F 1 ; (iv) фосфорилирование ATF-1, индуцированное PGF 2α , ослаблялось ингибиторами дыхательной цепи митохондрий; (v) индукция мРНК NOX1 с помощью PGF 2α , PDGF, FBS или PMA подавлялась интерференцией РНК, нацеленной на последовательность мРНК ATF-1; и (vi) сверхэкспрессия ATF-1 восстанавливала экспрессию NOX1, которая подавлялась олигомицином.
Ингибитор флавопротеинов DPI часто используется в качестве ингибитора НАДФН-оксидазы [8–10], поскольку он снижает активность НАДФН-оксидазы, образуя аддукты с восстановленным флавином [20]. Полученные данные позволяют предположить, что DPI является не только ингибитором фермента НАДФН-оксидазы, но также подавляющим фактором NOX1, каталитической субъединицы фермента. В последнем эффекте DPI было продемонстрировано участие митохондриальной дыхательной цепи.
Сообщается, что у того же члена семейства CREB / ATF CREB является одним из факторов транскрипции, активируемых митохондриальной дисфункцией.Arnould et al. [21] сообщили, что CREB участвует в сигнальном пути, который сообщает ядру состояние активности митохондрий. В клеточной линии, лишенной мтДНК (митохондриальной ДНК), CREB был конститутивно активирован фосфорилированием по Ser 133 , а метаболические ингибиторы митохондрий приводили к фосфорилированию CREB в эмбриональных клетках почек человека [21]. Напротив, метаболические ингибиторы митохондрий подавляли индуцированное PGF 2α фосфорилирование ATF-1 в клетках A7r5. Такое несоответствие во взаимосвязи между статусом фосфорилирования CREB / ATF и митохондриальной активностью может быть связано с различием в клеточном происхождении, используемом в этих исследованиях.
Такое несоответствие во взаимосвязи между статусом фосфорилирования CREB / ATF и митохондриальной активностью может быть связано с различием в клеточном происхождении, используемом в этих исследованиях.
ATF-1, но не CREB, фосфорилировался в клетках, стимулированных PGF 2α , хотя высокое сходство последовательностей обнаружено в сайтах, фосфорилированных серин / треонинкиназами. Это указывает на то, что существуют строгие механизмы, которые различают сайты фосфорилирования ATF-1 и CREB, а ATF-1 и CREB могут иметь разные функциональные роли в VSMC.Фактически, ATF-1, как сообщалось, участвует в индуцированном тромбином росте VSMC, тогда как CREB участвует в индуцированной ангиотензином-II гипертрофии VSMC [18,19]. Ранее мы сообщали, что индуцированная PGF 2α гипертрофия VSMC опосредуется индукцией мРНК NOX1 [7]. В свете настоящих открытий было ясно показано участие ATF-1 в PGF 2α -индуцированной гипертрофии VSMC. Интересно, что рецептор ангиотензина AT 1 , рецептор тромбина и простаноидный рецептор FP (PGF) являются рецепторами, связанными с G q / 11 .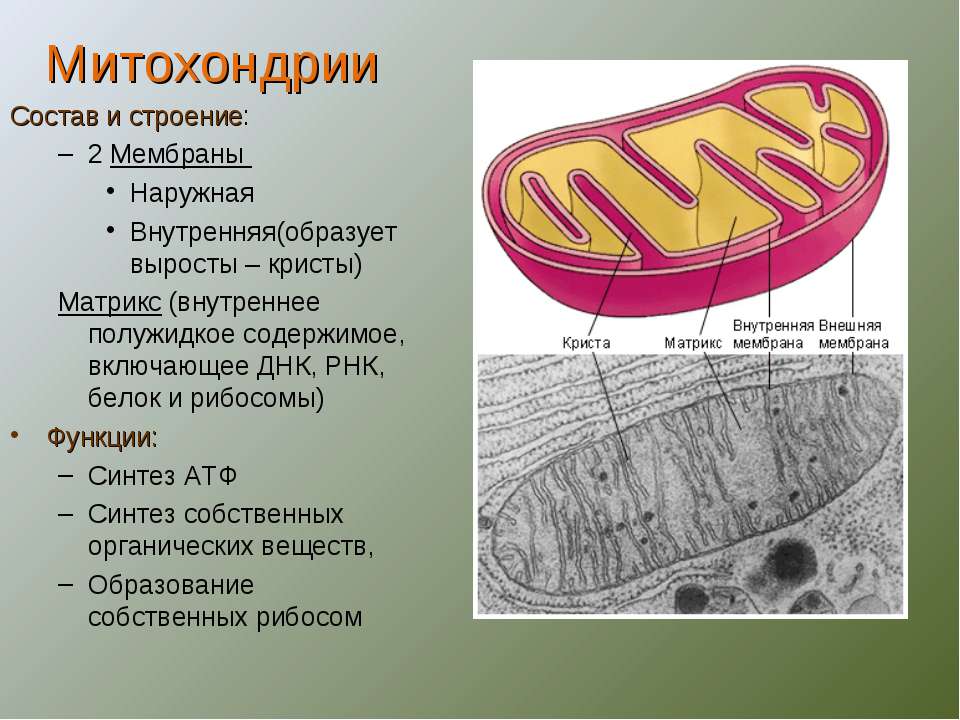 Эти рецепторы могут быть связаны с еще не идентифицированными активационными единицами, которые различают ATF-1 и CREB.
Эти рецепторы могут быть связаны с еще не идентифицированными активационными единицами, которые различают ATF-1 и CREB.
Молекулярные механизмы, с помощью которых PGF 2α — или PDGF-индуцированные сигналы распространяются к митохондриям и к фактору транскрипции ATF-1, все еще неизвестны. PGF 2α проявляет свое биологическое действие через связывание со своим специфическим рецептором FP на плазматических мембранах [22]. FP связан с оборотом фосфоинозитидов и последующей мобилизацией цитозольного Ca 2+ [23].PGF 2α также индуцирует транслокацию PKCε (протеинкиназы Cε) к мембране миоцитов [17]. С другой стороны, предыдущие исследования продемонстрировали, что PKCδ и PKCε нацелены на митохондрии после активации, а PKCδ изменяет потенциал митохондриальной мембраны [24–26]. Сообщается также, что PKCε совместно локализуется с ERK (киназа, регулируемая внеклеточными сигналами) в сердечных митохондриях и формирует сигнальные модули PKCε-ERK, которые принимают участие в PKCε-опосредованной кардиопротекции [27].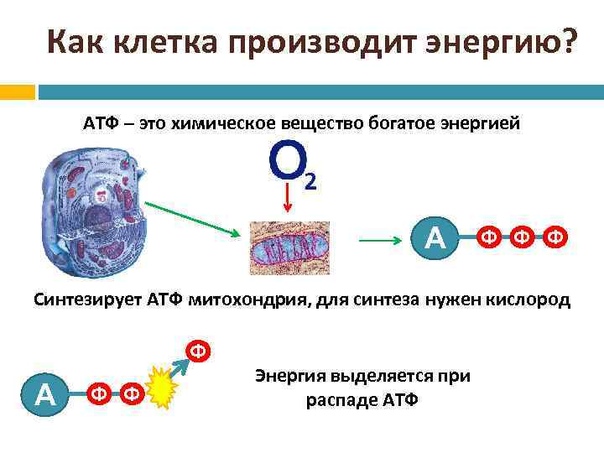 PDGF, эффекты которого опосредуются рецепторной тирозинкиназой, также, как сообщается, активирует ERK PKC-зависимым образом [28].В другом направлении исследований мы наблюдали, что индуцированная PGF 2α экспрессия мРНК NOX1 была значительно подавлена PD98059, ингибитором MEK1 / 2 (митоген-активируемая протеинкиназа / ERK-киназа 1/2), который блокирует путь ERK1 / 2. (Ч. Фан, М. Кацуяма, Т. Нишинака и Ч. Ябэ-Нисимура, неопубликованная работа). Поскольку о локализации CREB также сообщалось в митохондриях мозга [29], можно предположить, что эти PKC могут опосредовать PGF 2α — или PDGF-индуцированные сигналы в митохондрии, и что ERK и / или ATF-1, тесно связанные с митохондриями, могут играют ключевую роль в передаче сигналов в ядро, что приводит к экспрессии гена NOX1.Пока неясно, активирует ли ATF-1 прямо или косвенно транскрипцию NOX1. Элемент (ы), отвечающий за PGF 2α , не был обнаружен анализом люциферазного репортера с использованием конструкции, содержащей ок.
PDGF, эффекты которого опосредуются рецепторной тирозинкиназой, также, как сообщается, активирует ERK PKC-зависимым образом [28].В другом направлении исследований мы наблюдали, что индуцированная PGF 2α экспрессия мРНК NOX1 была значительно подавлена PD98059, ингибитором MEK1 / 2 (митоген-активируемая протеинкиназа / ERK-киназа 1/2), который блокирует путь ERK1 / 2. (Ч. Фан, М. Кацуяма, Т. Нишинака и Ч. Ябэ-Нисимура, неопубликованная работа). Поскольку о локализации CREB также сообщалось в митохондриях мозга [29], можно предположить, что эти PKC могут опосредовать PGF 2α — или PDGF-индуцированные сигналы в митохондрии, и что ERK и / или ATF-1, тесно связанные с митохондриями, могут играют ключевую роль в передаче сигналов в ядро, что приводит к экспрессии гена NOX1.Пока неясно, активирует ли ATF-1 прямо или косвенно транскрипцию NOX1. Элемент (ы), отвечающий за PGF 2α , не был обнаружен анализом люциферазного репортера с использованием конструкции, содержащей ок. 2 т.п.н. 5′-фланкирующей области гена NOX1 мыши (результаты не показаны). Дальнейшие исследования промотора и энхансера гена NOX1 должны привести к лучшему пониманию регуляции экспрессии гена NOX1 в VSMC. Настоящее исследование предоставляет первые доказательства роли митохондрий и ATF-1, члена семейства CREB / ATF, в регуляции экспрессии гена NOX1.
2 т.п.н. 5′-фланкирующей области гена NOX1 мыши (результаты не показаны). Дальнейшие исследования промотора и энхансера гена NOX1 должны привести к лучшему пониманию регуляции экспрессии гена NOX1 в VSMC. Настоящее исследование предоставляет первые доказательства роли митохондрий и ATF-1, члена семейства CREB / ATF, в регуляции экспрессии гена NOX1.
Мультимедийное дополнение
Дополнительные рисунки 1 и 2:Благодарности
Эта работа была частично поддержана грантом для молодых ученых (B) 14770036 Министерства образования, культуры, спорта, науки и технологий Японии. Мы благодарим доктора Э. Фунакоши с факультета фармацевтических наук Университета Сецунан, Хираката, Осака, Япония, за ценные обсуждения и советы.
Список литературы
1. Гриндлинг К. К., Сореску Д., Ушио-Фукаи М. NAD (P) H оксидаза: роль в сердечно-сосудистой биологии и болезнях. Circ. Res. 2000; 86: 494–501. [PubMed] [Google Scholar] 2. Гузик Т. Дж., Вест Н. Е., Блэк Э.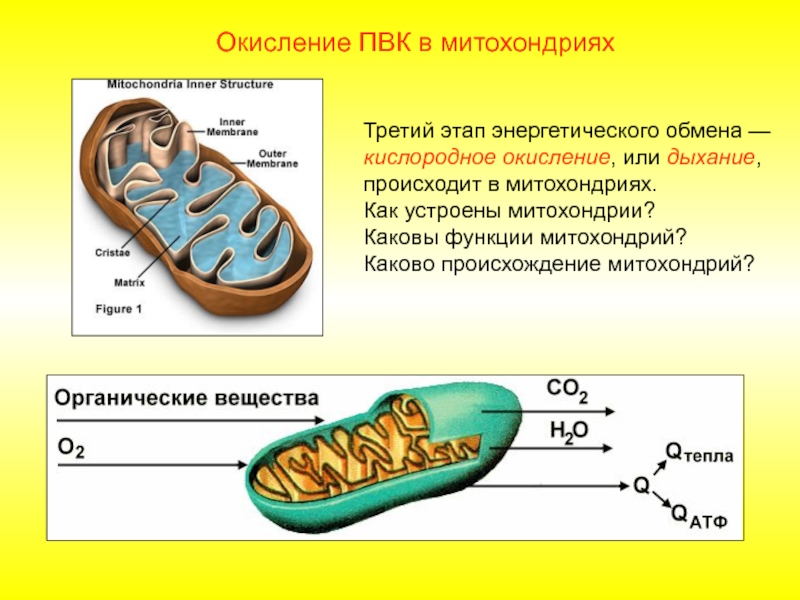 , Макдональд Д., Ратнатунга К., Пиллай Р., Чаннон К. М. Производство сосудистого супероксида НАД (Ф) Н-оксидазой: связь с эндотелиальной дисфункцией и клинические проявления. факторы риска. Circ. Res. 2000; 86: E85 – E90. [PubMed] [Google Scholar] 3. Ирани К. Передача сигналов окислителя в росте, гибели и выживании сосудистых клеток: обзор роли активных форм кислорода в митогенных и апоптотических сигналах гладких мышц и эндотелиальных клеток.Circ. Res. 2000. 87: 179–183. [PubMed] [Google Scholar] 4. Лассег Б., Клемпус Р. Э. Сосудистые NAD (P) H оксидазы: особенности, экспрессия и регуляция. Являюсь. J. Physiol. Regul. Интегр. Комп. Physiol. 2003; 285: R277 – R297. [PubMed] [Google Scholar] 5. Сух Я. А., Арнольд Р. С., Лассег Б., Ши Дж., Сюй Х., Сореску Д., Чанг А. Б., Гриндлинг К. К., Ламбет Дж. Д. Трансформация клеток супероксидом -генерирующая оксидаза Mox1. Nature (Лондон) 1999; 401: 79–82. [PubMed] [Google Scholar] 6. Лассег Б., Сореску Д., Szöcs K., Yin Q., Akers M., Zhang Y., Grant S.
, Макдональд Д., Ратнатунга К., Пиллай Р., Чаннон К. М. Производство сосудистого супероксида НАД (Ф) Н-оксидазой: связь с эндотелиальной дисфункцией и клинические проявления. факторы риска. Circ. Res. 2000; 86: E85 – E90. [PubMed] [Google Scholar] 3. Ирани К. Передача сигналов окислителя в росте, гибели и выживании сосудистых клеток: обзор роли активных форм кислорода в митогенных и апоптотических сигналах гладких мышц и эндотелиальных клеток.Circ. Res. 2000. 87: 179–183. [PubMed] [Google Scholar] 4. Лассег Б., Клемпус Р. Э. Сосудистые NAD (P) H оксидазы: особенности, экспрессия и регуляция. Являюсь. J. Physiol. Regul. Интегр. Комп. Physiol. 2003; 285: R277 – R297. [PubMed] [Google Scholar] 5. Сух Я. А., Арнольд Р. С., Лассег Б., Ши Дж., Сюй Х., Сореску Д., Чанг А. Б., Гриндлинг К. К., Ламбет Дж. Д. Трансформация клеток супероксидом -генерирующая оксидаза Mox1. Nature (Лондон) 1999; 401: 79–82. [PubMed] [Google Scholar] 6. Лассег Б., Сореску Д., Szöcs K., Yin Q., Akers M., Zhang Y., Grant S. L., Lambeth J. D., Griendling K. K. Новые гомологи gp91 phox в гладкомышечных клетках сосудов: NOX1 опосредует ангиотензин II -индуцированное образование супероксида и редокс-чувствительные сигнальные пути. Circ. Res. 2001; 88: 888–894. [PubMed] [Google Scholar] 7. Katsuyama M., Fan C., Yabe-Nishimura C. НАДФН-оксидаза участвует в индуцированной простагландином F 2α гипертрофии гладкомышечных клеток сосудов: индукция NOX1 с помощью PGF 2α J.Биол. Chem. 2002; 277: 13438–13442. [PubMed] [Google Scholar] 8. Эллис Дж. А., Майер С. Дж., Джонс О. Т. Влияние ингибитора НАДФН-оксидазы дифенилениодония на аэробную и анаэробную микробицидную активность нейтрофилов человека. Биохим. J. 1988; 251: 887–891. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9. Робертсон А. К., Кросс А. Р., Джонс О. Т., Эндрю П. В. Использование дифенилен-йодония, ингибитора НАДФН-оксидазы, для исследования антимикробного действия макрофагов, полученных из моноцитов человека.J. Immunol.
L., Lambeth J. D., Griendling K. K. Новые гомологи gp91 phox в гладкомышечных клетках сосудов: NOX1 опосредует ангиотензин II -индуцированное образование супероксида и редокс-чувствительные сигнальные пути. Circ. Res. 2001; 88: 888–894. [PubMed] [Google Scholar] 7. Katsuyama M., Fan C., Yabe-Nishimura C. НАДФН-оксидаза участвует в индуцированной простагландином F 2α гипертрофии гладкомышечных клеток сосудов: индукция NOX1 с помощью PGF 2α J.Биол. Chem. 2002; 277: 13438–13442. [PubMed] [Google Scholar] 8. Эллис Дж. А., Майер С. Дж., Джонс О. Т. Влияние ингибитора НАДФН-оксидазы дифенилениодония на аэробную и анаэробную микробицидную активность нейтрофилов человека. Биохим. J. 1988; 251: 887–891. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9. Робертсон А. К., Кросс А. Р., Джонс О. Т., Эндрю П. В. Использование дифенилен-йодония, ингибитора НАДФН-оксидазы, для исследования антимикробного действия макрофагов, полученных из моноцитов человека.J. Immunol.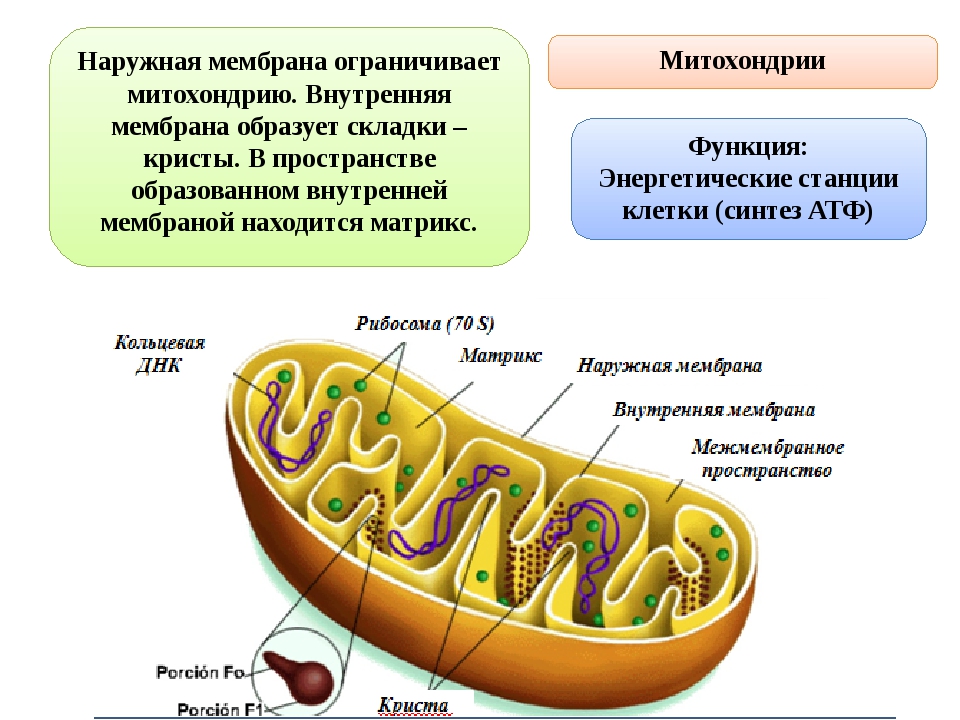 Методы. 1990; 133: 175–179. [PubMed] [Google Scholar] 10. Doussiere J., Vignais P. V. Дифенилен-йодоний как ингибитор НАДФН-оксидазного комплекса бычьих нейтрофилов: факторы, контролирующие ингибирующую способность дифенилен-йодония в бесклеточной системе активации оксидазы. Евро. J. Biochem. 1992; 208: 61–71. [PubMed] [Google Scholar] 11. Nishinaka T., Yabe-Nishimura C. Путь рецептора EGF-ERK является основным сигнальным путем, который опосредует повышающую регуляцию экспрессии альдозоредуктазы при окислительном стрессе.Free Radical Biol. Med. 2001; 31: 205–216. [PubMed] [Google Scholar] 12. Эндрюс Н. С., Фаллер Д. В. Быстрый метод микропрепаратов для извлечения ДНК-связывающих белков из ограниченного числа клеток млекопитающих. Nucleic Acids Res. 1991; 19: 2499. [Бесплатная статья PMC] [PubMed] [Google Scholar] 13. Miyagishi M., Taira K. Управляемые промотором U6 миРНК с четырьмя уридиновыми 3′-выступами эффективно подавляют экспрессию целевых генов в клетках млекопитающих. Nat. Biotechnol.
Методы. 1990; 133: 175–179. [PubMed] [Google Scholar] 10. Doussiere J., Vignais P. V. Дифенилен-йодоний как ингибитор НАДФН-оксидазного комплекса бычьих нейтрофилов: факторы, контролирующие ингибирующую способность дифенилен-йодония в бесклеточной системе активации оксидазы. Евро. J. Biochem. 1992; 208: 61–71. [PubMed] [Google Scholar] 11. Nishinaka T., Yabe-Nishimura C. Путь рецептора EGF-ERK является основным сигнальным путем, который опосредует повышающую регуляцию экспрессии альдозоредуктазы при окислительном стрессе.Free Radical Biol. Med. 2001; 31: 205–216. [PubMed] [Google Scholar] 12. Эндрюс Н. С., Фаллер Д. В. Быстрый метод микропрепаратов для извлечения ДНК-связывающих белков из ограниченного числа клеток млекопитающих. Nucleic Acids Res. 1991; 19: 2499. [Бесплатная статья PMC] [PubMed] [Google Scholar] 13. Miyagishi M., Taira K. Управляемые промотором U6 миРНК с четырьмя уридиновыми 3′-выступами эффективно подавляют экспрессию целевых генов в клетках млекопитающих. Nat. Biotechnol. 2002; 20: 497–500. [PubMed] [Google Scholar] 14.Йокота Т., Сакамото Н., Эномото Н., Танабэ Ю., Миягиши М., Маэкава С., Йи Л., Куросаки М., Тайра К., Ватанабэ М., Мизусава Х. Ингибирование внутриклеточной репликации вируса гепатита С. синтетическими и полученными из вектора малыми интерферирующими РНК. EMBO Rep. 2003; 4: 602–608. [Бесплатная статья PMC] [PubMed] [Google Scholar] 15. Стер Д. Дж., Фасехун О. А., Квон Н. С., Гросс С. С., Гонсалес Дж. А., Леви Р., Натан К. Ф. Ингибирование синтазы оксида азота макрофагов и эндотелиальных клеток дифенилениодонием и его аналоги.FASEB J. 1991; 5: 98–103. [PubMed] [Google Scholar] 16. Ли Ю., Труш М. А. Дифенилениодоний, ингибитор НАД (Ф) Н-оксидазы, также сильно подавляет выработку митохондриальных активных форм кислорода. Биохим. Биофиз. Res. Commun. 1998. 253: 295–299. [PubMed] [Google Scholar] 17. Адамс Дж. У., Сах В. П., Хендерсон С. А., Браун Дж. Х. Тирозинкиназа и c-Jun NH 2 -концевая киназа опосредуют гипертрофические ответы на простагландин F 2α в культивируемых желудочковых клетках новорожденных крыс.
2002; 20: 497–500. [PubMed] [Google Scholar] 14.Йокота Т., Сакамото Н., Эномото Н., Танабэ Ю., Миягиши М., Маэкава С., Йи Л., Куросаки М., Тайра К., Ватанабэ М., Мизусава Х. Ингибирование внутриклеточной репликации вируса гепатита С. синтетическими и полученными из вектора малыми интерферирующими РНК. EMBO Rep. 2003; 4: 602–608. [Бесплатная статья PMC] [PubMed] [Google Scholar] 15. Стер Д. Дж., Фасехун О. А., Квон Н. С., Гросс С. С., Гонсалес Дж. А., Леви Р., Натан К. Ф. Ингибирование синтазы оксида азота макрофагов и эндотелиальных клеток дифенилениодонием и его аналоги.FASEB J. 1991; 5: 98–103. [PubMed] [Google Scholar] 16. Ли Ю., Труш М. А. Дифенилениодоний, ингибитор НАД (Ф) Н-оксидазы, также сильно подавляет выработку митохондриальных активных форм кислорода. Биохим. Биофиз. Res. Commun. 1998. 253: 295–299. [PubMed] [Google Scholar] 17. Адамс Дж. У., Сах В. П., Хендерсон С. А., Браун Дж. Х. Тирозинкиназа и c-Jun NH 2 -концевая киназа опосредуют гипертрофические ответы на простагландин F 2α в культивируемых желудочковых клетках новорожденных крыс.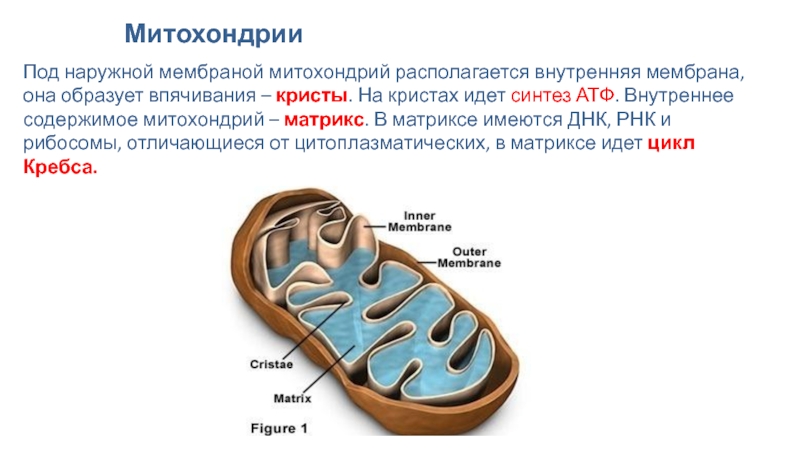 миоциты. Circ.Res. 1998. 83: 167–178. [PubMed] [Google Scholar] 18. Фунакоши Ю., Ичики Т., Такеда К., Токуно Т., Иино Н., Такешита А. Критическая роль белка, связывающего элемент цАМФ-ответа, в индуцированной ангиотензином II гипертрофии гладкомышечных клеток сосудов. J. Biol. Chem. 2002; 277: 18710–18717. [PubMed] [Google Scholar] 19. Гош С. К., Гадипарти Л., Зенг З. З., Бханори М., Теллез К., Бар-Эли М., Рао Г. Н. ATF-1 опосредует активируемый протеазой рецептор-1, но не рецепторную тирозинкиназу -индуцированный синтез ДНК в гладкомышечных клетках сосудов.J. Biol. Chem. 2002; 277: 21325–21331. [PubMed] [Google Scholar] 20. О’Доннелл Б. В., Тью Д. Г., Джонс О. Т., Англия П. Дж. Исследования механизма ингибирования йодониевых соединений с особым упором на НАДФН-оксидазу нейтрофилов. Биохим. J. 1993; 290: 41–49. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C., Remacle J., Raes M. Активация CREB, вызванная дисфункцией митохондрий, является новым сигнальным путем, который нарушает пролиферацию клеток.
миоциты. Circ.Res. 1998. 83: 167–178. [PubMed] [Google Scholar] 18. Фунакоши Ю., Ичики Т., Такеда К., Токуно Т., Иино Н., Такешита А. Критическая роль белка, связывающего элемент цАМФ-ответа, в индуцированной ангиотензином II гипертрофии гладкомышечных клеток сосудов. J. Biol. Chem. 2002; 277: 18710–18717. [PubMed] [Google Scholar] 19. Гош С. К., Гадипарти Л., Зенг З. З., Бханори М., Теллез К., Бар-Эли М., Рао Г. Н. ATF-1 опосредует активируемый протеазой рецептор-1, но не рецепторную тирозинкиназу -индуцированный синтез ДНК в гладкомышечных клетках сосудов.J. Biol. Chem. 2002; 277: 21325–21331. [PubMed] [Google Scholar] 20. О’Доннелл Б. В., Тью Д. Г., Джонс О. Т., Англия П. Дж. Исследования механизма ингибирования йодониевых соединений с особым упором на НАДФН-оксидазу нейтрофилов. Биохим. J. 1993; 290: 41–49. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C., Remacle J., Raes M. Активация CREB, вызванная дисфункцией митохондрий, является новым сигнальным путем, который нарушает пролиферацию клеток. EMBO J. 2002; 21: 53–63. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22. Сугимото Ю., Хасумото К., Намба Т., Ирие А., Кацуяма М., Негиси М., Какидзука А., Нарумия С., Итикава А. Клонирование и экспрессия кДНК для рецептора простагландина F мыши. J. Biol. Chem. 1994; 269: 1356–1360. [PubMed] [Google Scholar] 23. Гриффин Б. В., Магнино П. Е., Панг И. Х., Шариф Н. А. Фармакологическая характеристика рецептора простагландина FP на гладкомышечных клетках сосудов крыс (A7r5), связанных с обменом фосфоинозитидов и мобилизацией внутриклеточного кальция.J. Pharmacol. Exp. Ther. 1998. 286: 411–418. [PubMed] [Google Scholar] 24. Ли Л., Лоренцо П. С., Боги К., Блумберг П. М., Юспа С. Х. Протеинкиназа Cδ нацелена на митохондрии, изменяет потенциал митохондриальной мембраны и индуцирует апоптоз в нормальных и неопластических кератиноцитах при сверхэкспрессии аденовирусным вектором . Мол. Клетка. Биол. 1999; 19: 8547–8558. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Фрайер Р. М., Ван Ю.
EMBO J. 2002; 21: 53–63. [Бесплатная статья PMC] [PubMed] [Google Scholar] 22. Сугимото Ю., Хасумото К., Намба Т., Ирие А., Кацуяма М., Негиси М., Какидзука А., Нарумия С., Итикава А. Клонирование и экспрессия кДНК для рецептора простагландина F мыши. J. Biol. Chem. 1994; 269: 1356–1360. [PubMed] [Google Scholar] 23. Гриффин Б. В., Магнино П. Е., Панг И. Х., Шариф Н. А. Фармакологическая характеристика рецептора простагландина FP на гладкомышечных клетках сосудов крыс (A7r5), связанных с обменом фосфоинозитидов и мобилизацией внутриклеточного кальция.J. Pharmacol. Exp. Ther. 1998. 286: 411–418. [PubMed] [Google Scholar] 24. Ли Л., Лоренцо П. С., Боги К., Блумберг П. М., Юспа С. Х. Протеинкиназа Cδ нацелена на митохондрии, изменяет потенциал митохондриальной мембраны и индуцирует апоптоз в нормальных и неопластических кератиноцитах при сверхэкспрессии аденовирусным вектором . Мол. Клетка. Биол. 1999; 19: 8547–8558. [Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Фрайер Р. М., Ван Ю. , Хсу А. К., Гросс Г. Дж. Существенная активация PKC-δ в инициируемой опиоидами кардиопротекции.Являюсь. J. Physiol. Heart Circ. Physiol. 2001; 280: h2346 – h2353. [PubMed] [Google Scholar] 26. Маджумдер П. К., Мишра Н. С., Сан X., Бхарти А., Харбанда С., Саксена С., Куфе Д. Направление протеинкиназы Cδ в митохондрии в ответ на окислительный стресс. Рост клеток различается. 2001; 12: 465–470. [PubMed] [Google Scholar] 27. Baines C.P., Zhang J., Wang G. W., Zheng Y. T., Xiu J. X., Cardwell E. M., Bolli R., Ping P. Митохондриальные PKCε и MAPK образуют сигнальные модули в сердце мыши: усиление митохондриальных взаимодействий PKCε – MAPK и дифференциальная активация MAPK в PKCε-индуцированной кардиопротекции.Circ. Res. 2002; 90: 390–397. [PubMed] [Google Scholar] 28. Tsakiridis T., Tsiani E., Lekas P., Bergman A., Cherepanov V., Whiteside C., Downey G.P. Инсулин, инсулиноподобный фактор роста-I и тромбоцитарный фактор роста активируют внеклеточные сигналы, регулируемые киназы различными путями в мышечных клетках.
, Хсу А. К., Гросс Г. Дж. Существенная активация PKC-δ в инициируемой опиоидами кардиопротекции.Являюсь. J. Physiol. Heart Circ. Physiol. 2001; 280: h2346 – h2353. [PubMed] [Google Scholar] 26. Маджумдер П. К., Мишра Н. С., Сан X., Бхарти А., Харбанда С., Саксена С., Куфе Д. Направление протеинкиназы Cδ в митохондрии в ответ на окислительный стресс. Рост клеток различается. 2001; 12: 465–470. [PubMed] [Google Scholar] 27. Baines C.P., Zhang J., Wang G. W., Zheng Y. T., Xiu J. X., Cardwell E. M., Bolli R., Ping P. Митохондриальные PKCε и MAPK образуют сигнальные модули в сердце мыши: усиление митохондриальных взаимодействий PKCε – MAPK и дифференциальная активация MAPK в PKCε-индуцированной кардиопротекции.Circ. Res. 2002; 90: 390–397. [PubMed] [Google Scholar] 28. Tsakiridis T., Tsiani E., Lekas P., Bergman A., Cherepanov V., Whiteside C., Downey G.P. Инсулин, инсулиноподобный фактор роста-I и тромбоцитарный фактор роста активируют внеклеточные сигналы, регулируемые киназы различными путями в мышечных клетках. Биохим. Биофиз. Res. Commun. 2001. 288: 205–211. [PubMed] [Google Scholar] 29. Каммарота М., Паратча Г., Бевилаква Л. Р., Леви де Стейн М., Лопес М., Пеллегрино де Иральди А., Искьердо И., Медина Дж.H. Циклический AMP-чувствительный элемент, связывающий белок в митохондриях мозга. J. Neurochem. 1999; 72: 2272–2277. [PubMed] [Google Scholar]
Биохим. Биофиз. Res. Commun. 2001. 288: 205–211. [PubMed] [Google Scholar] 29. Каммарота М., Паратча Г., Бевилаква Л. Р., Леви де Стейн М., Лопес М., Пеллегрино де Иральди А., Искьердо И., Медина Дж.H. Циклический AMP-чувствительный элемент, связывающий белок в митохондриях мозга. J. Neurochem. 1999; 72: 2272–2277. [PubMed] [Google Scholar] Роль пути ISR-ATF4 и его перекрестная связь с Nrf2 в контроле качества митохондрий
J Clin Biochem Nutr. 2019 Янв; 64 (1): 1–12.
Шуя Касаи
1 Департамент науки о стресс-реагировании, Центр перспективных медицинских исследований, Высшая школа медицины Университета Хиросаки, 5 Дзайфу-чо, Хиросаки 036-8562, Япония
Хироми Ямадзаки
1 Департамент Наука о реакции на стресс, Центр передовых медицинских исследований, Высшая школа медицины Университета Хиросаки, 5 Зайфу-тё, Хиросаки 036-8562, Япония
Куниказу Танджи
2 Отделение невропатологии Высшей школы медицины Университета Хиросаки, 5 Дзайфу -cho, Hirosaki 036-8562, Япония
Máté János Engler
1 Департамент науки о стресс-реагировании, Центр передовых медицинских исследований, Высшая школа медицины Университета Хиросаки, 5 Zaifu-cho, Hirosaki 036-8562, Япония
Tomoh Matsumiya
3 Кафедра сосудистой биологии, Высшая школа медицины Университета Хиросаки, 5 Zaifu-cho, Hirosaki 036-85 62, Япония
Кен Ито
1 Департамент науки о реакции на стресс, Центр передовых медицинских исследований, Высшая школа медицины Университета Хиросаки, 5 Дзайфу-чо, Хиросаки 036-8562, Япония
1 Департамент стресса Response Science, Центр передовых медицинских исследований, Высшая школа медицины Университета Хиросаки, 5 Zaifu-cho, Hirosaki 036-8562, Япония
2 Отделение невропатологии Высшей школы медицины Университета Хиросаки, 5 Zaifu-cho, Hirosaki 036 -8562, Япония
3 Кафедра сосудистой биологии, Высшая школа медицины Университета Хиросаки, 5 Zaifu-cho, Hirosaki 036-8562, Япония
Поступила в редакцию 22 марта 2018 г . ; Принят в печать 11 июня 2018 г.
; Принят в печать 11 июня 2018 г.
Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии правильного цитирования оригинальной работы.
Эта статья цитируется в других статьях в PMC.Abstract
Недавние исследования прояснили важность митохондрий в различных возрастных дегенеративных заболеваниях, включая болезнь Альцгеймера с поздним началом и болезнь Паркинсона.Хотя митохондриальные нарушения могут быть вовлечены в каждый этап прогрессирования заболевания, несколько наблюдений продемонстрировали, что незначительные функциональные нарушения митохондрий наблюдаются до фактического появления патофизиологических изменений и могут быть целью раннего терапевтического вмешательства. Сигналы от поврежденных митохондрий передаются в ядро, что приводит к измененной экспрессии кодируемых ядром генов, в том числе митохондриальных белков (т.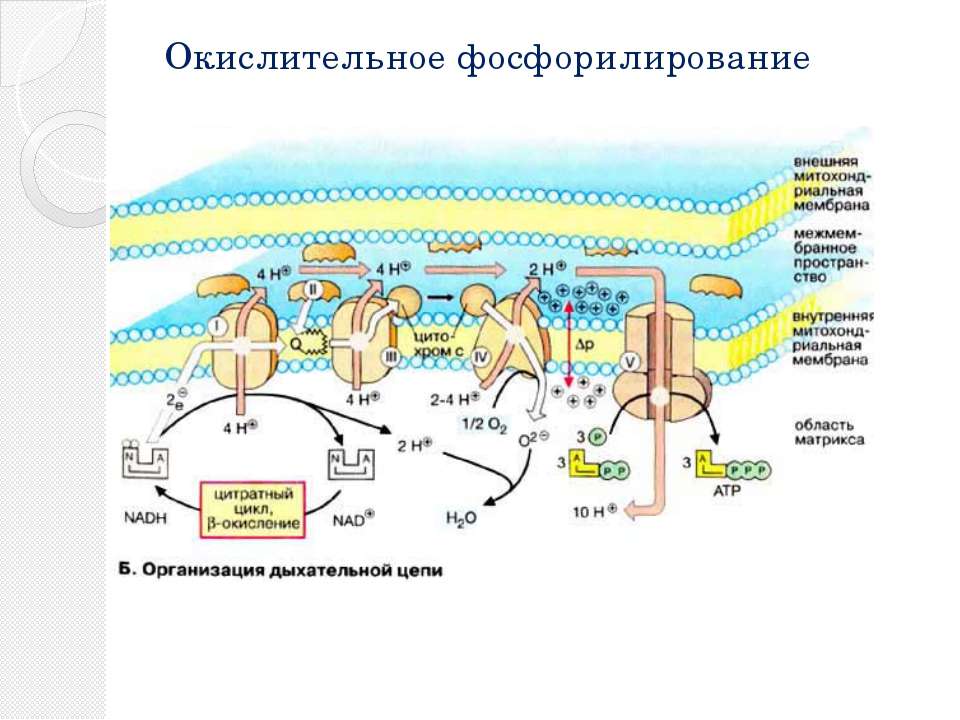 е. митохондриальной ретроградной передачи сигналов).Митохондриальная ретроградная передача сигналов улучшает митохондриальные пертурбации (например, митогормезис) и считается гомеостатической стрессовой реакцией на внутренние (например, старение или патологические мутации) и внешние (например, химические вещества и патогены) стимулы. Существует несколько ветвей митохондриальной ретроградной передачи сигналов, включая митохондриальный ответ развернутого белка (UPR MT ), но недавние наблюдения все больше показывают важность пути ISR-ATF4 в митохондриальной ретроградной передаче сигналов.Кроме того, Nrf2, главный регулятор реакции на окислительный стресс, взаимодействует с ATF4 и кооперативно активирует батарею антиоксидантных и антиапоптотических генов, репрессируя опосредованный ATF4 проапоптотический ген, CHOP. В этой обзорной статье мы суммировали восходящие и нисходящие механизмы активации ATF4 во время митохондриальных стрессов и нарушений и обсуждали терапевтическое вмешательство против дегенеративных заболеваний с использованием активаторов Nrf2.
е. митохондриальной ретроградной передачи сигналов).Митохондриальная ретроградная передача сигналов улучшает митохондриальные пертурбации (например, митогормезис) и считается гомеостатической стрессовой реакцией на внутренние (например, старение или патологические мутации) и внешние (например, химические вещества и патогены) стимулы. Существует несколько ветвей митохондриальной ретроградной передачи сигналов, включая митохондриальный ответ развернутого белка (UPR MT ), но недавние наблюдения все больше показывают важность пути ISR-ATF4 в митохондриальной ретроградной передаче сигналов.Кроме того, Nrf2, главный регулятор реакции на окислительный стресс, взаимодействует с ATF4 и кооперативно активирует батарею антиоксидантных и антиапоптотических генов, репрессируя опосредованный ATF4 проапоптотический ген, CHOP. В этой обзорной статье мы суммировали восходящие и нисходящие механизмы активации ATF4 во время митохондриальных стрессов и нарушений и обсуждали терапевтическое вмешательство против дегенеративных заболеваний с использованием активаторов Nrf2.
Ключевые слова: ATF4, Nrf2, митохондрии, GCN2
Введение
С момента поглощения α-протеобактерий археями примерно 1.5 миллиардов лет назад общение между двумя сущностями имело решающее значение в зарождении и развитии эукариот. (1) По мере того как α-протеобактерии трансформируются в текущие митохондрии, выработка энергии эукариотами все больше ограничивается митохондриями за счет окислительного фосфорилирования (OxPhos). В контексте современных эукариот важна мито-ядерная коммуникация, особенно потому, что белки OxPhos в митохондриях представляют собой мозаику белков, кодируемых ядром и митохондриями, за исключением комплекса II, а белки, кодируемые митохондриями, действуют как ядро сборки белков OxPhos. и должны быть строго согласованы с эффективностью OxPhos. (2) Первоначально митохондрии могут использовать локально генерируемые активные формы кислорода (АФК) в качестве сигнала внутри митохондрий в ответ на повышенную потребность в энергии для изменения содержания и сборки митохондриальной электронной транспортной цепи (mETC) или для увеличения митохондриального биогенеза.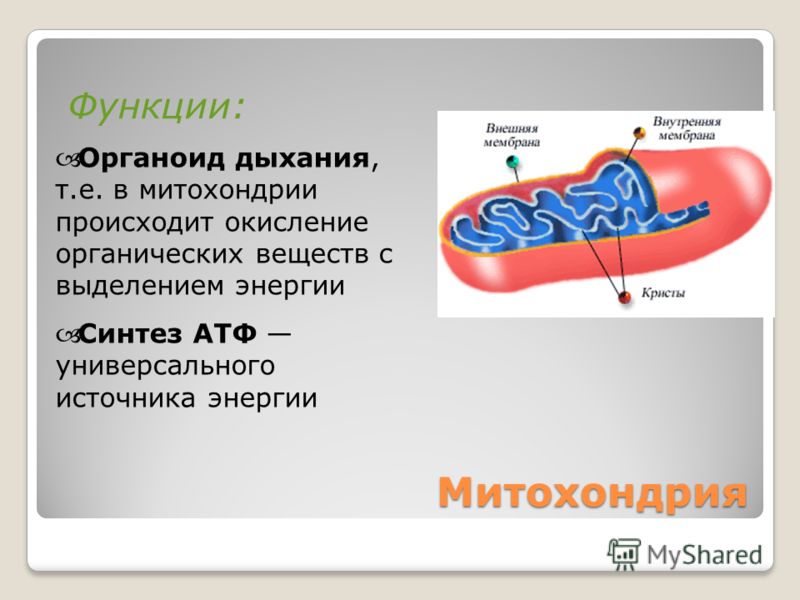 (т.е. реактивный биогенез). (1,3) Пока еще неопределенные сигналы организуют скоординированный биосинтез митохондриальных и ядерно-кодируемых белков, чтобы максимизировать эффективность OxPhos.Недавнее сообщение о дрожжах продемонстрировало, что регуляторный механизм трансляции управляет сбалансированным синтезом белков OxPhos, кодируемых митохондриями и ядром, во время энергетического сдвига от гликолиза к OxPhos. (4,5)
(т.е. реактивный биогенез). (1,3) Пока еще неопределенные сигналы организуют скоординированный биосинтез митохондриальных и ядерно-кодируемых белков, чтобы максимизировать эффективность OxPhos.Недавнее сообщение о дрожжах продемонстрировало, что регуляторный механизм трансляции управляет сбалансированным синтезом белков OxPhos, кодируемых митохондриями и ядром, во время энергетического сдвига от гликолиза к OxPhos. (4,5)
Ретроградная передача сигналов митохондрий (MRS), которая была первоначально описана у дрожжей, включает в себя множество механизмов, которые сигнализируют о митохондриальных аномалиях в ядро и вызывают антероградные механизмы (т. Е. От ядра к механизмам митохондрий. ), которые действуют для защиты от этих аномалий.У млекопитающих MRS включает ответ на митохондриальный развернутый белок (UPR MT ), энергетический стресс (6,7) , (8) Ca 2+ , высвобождение (9) и продукцию ROS ( 10) (рис.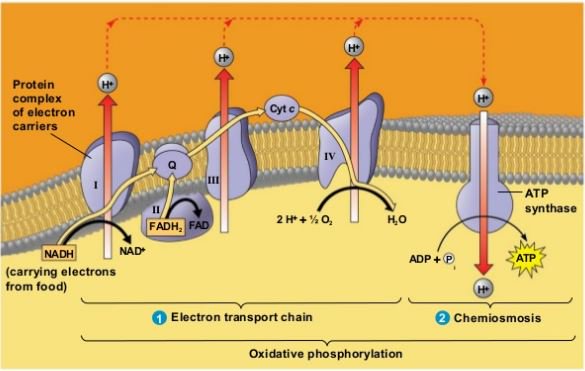 ). В этом обзоре, после первого описания общего обзора MRS дрожжей и млекопитающих, мы сосредоточимся на роли сигнального пути ATF (активирующий фактор транскрипции) 4 в контроле качества и гомеостазе митохондрий у видов млекопитающих (рис.).
). В этом обзоре, после первого описания общего обзора MRS дрожжей и млекопитающих, мы сосредоточимся на роли сигнального пути ATF (активирующий фактор транскрипции) 4 в контроле качества и гомеостазе митохондрий у видов млекопитающих (рис.).
Ретроградные сигнальные пути от митохондрий к ядру у млекопитающих. Митохондриальное дыхание производит собственные АФК, которые устраняются эндогенными антиоксидантами. Различные стрессы производят вредный избыток АФК и вызывают митохондриальную дисфункцию, которая приводит к снижению синтеза АТФ и митохондриальных метаболитов, потере митохондриального мембранного потенциала и высвобождению Са 2+ , накоплению развернутого белка и дальнейшему производству АФК. Энергетический стресс активирует AMPK и индуцирует экспрессию PGC1α- и FOXO-регулируемых генов, которые участвуют в митохондриальном биогенезе и репликации мтДНК. (107,108) Повышенный цитоплазматический Ca 2+ стимулирует гликолиз и захват Ca 2+ за счет активации CaMK и Cn. CaMK активирует экспрессию генов за счет фосфорилирования CREB и за счет активации AMPK-регулируемого пути, тогда как Cn активирует экспрессию NF-κB- и NFAT-зависимых генов. Кроме того, истощение мтДНК, ингибирование митохондриальных шаперонов или ингибирование митохондриальной протеазы индуцируют экспрессию митохондриальных шаперонинов, известных как UPR MT , у нематод, хотя молекулярный механизм у млекопитающих полностью не выяснен.ATF4 активируется в ответ на выработку митохондриальных АФК через PERK и / или GCN2 для усиления экспрессии шаперона, синтеза аминокислот и синтеза антиоксидантов. Повышенная цитоплазматическая АФК активирует Nrf2 и индуцирует экспрессию антиоксидантного гена. AMPK, AMP-активированная протеинкиназа; ATF4, активирующий фактор транскрипции 4; CaMK, кальмодулинкиназа; Cn, кальциневрин; CREB, белок, связывающий элемент ответа цАМФ; FOXO, вилочная коробка O; NFAT, ядерный фактор активированных Т-клеток; NF-κB, ядерный фактор-κB; PGC1α, коактиватор PPARγ 1α; АФК, активные формы кислорода.
CaMK активирует экспрессию генов за счет фосфорилирования CREB и за счет активации AMPK-регулируемого пути, тогда как Cn активирует экспрессию NF-κB- и NFAT-зависимых генов. Кроме того, истощение мтДНК, ингибирование митохондриальных шаперонов или ингибирование митохондриальной протеазы индуцируют экспрессию митохондриальных шаперонинов, известных как UPR MT , у нематод, хотя молекулярный механизм у млекопитающих полностью не выяснен.ATF4 активируется в ответ на выработку митохондриальных АФК через PERK и / или GCN2 для усиления экспрессии шаперона, синтеза аминокислот и синтеза антиоксидантов. Повышенная цитоплазматическая АФК активирует Nrf2 и индуцирует экспрессию антиоксидантного гена. AMPK, AMP-активированная протеинкиназа; ATF4, активирующий фактор транскрипции 4; CaMK, кальмодулинкиназа; Cn, кальциневрин; CREB, белок, связывающий элемент ответа цАМФ; FOXO, вилочная коробка O; NFAT, ядерный фактор активированных Т-клеток; NF-κB, ядерный фактор-κB; PGC1α, коактиватор PPARγ 1α; АФК, активные формы кислорода.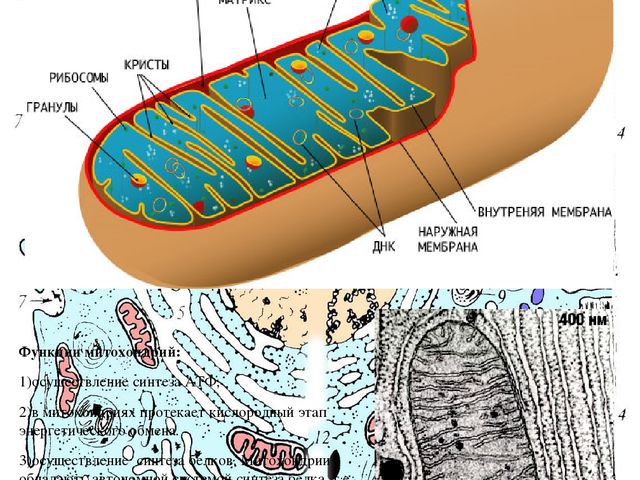
Обзор MRS в дрожжах
Исторически MRS широко изучалась и описывалась на дрожжах. Дрожжи могут выжить и размножаться без митохондриальной ДНК (т. Е. Ρ 0 клеток) при культивировании в среде, богатой ферментируемыми источниками углерода, такими как глюкоза. В дрожжевых клетках с дефектом дыхания, таких как клетки ρ 0 , MRS используется для корректировки потребности в метаболизме углерода и азота. (11) В этих клетках поток цикла TCA уменьшается, потому что реакция сукцинатдегидрогеназы не может протекать в отсутствие интактного mETC.В дрожжах, промежуточное звено цикла TCA, α-кетоглутарат, важен для внутриклеточного синтеза глутамата, который вместе с глутамином используется в качестве предшественника нуклеотидов и других аминокислот, обеспечивающих практически весь источник азота для биосинтетических нужд. (12) Для пополнения запасов α-кетоглутарата в клетках, клетки ρ 0 индуцируют массив генов анаплеротического пути, который включает в себя устойчиво индуцированные гены пероксисомального цикла глиоксилата (рис. ). (13) По существу, ингибирование MRS в клетках ρ 0 делает дрожжевые ауксотрофы глутамата. CIT2 , ген, важный для пероксисомального глиоксилатного цикла, и несколько других генов-мишеней MRS контролируются Rtg1p, Rtg2p и Rtg3p (13,14) (далее называемый путем RTG). Rtg2p необходим для активации факторов транскрипции bHLH-Zip, Rtg1p и Rtg3p в клетках ρ 0 . Отмеченная повышающая регуляция гена CIT2 , канонического гена-мишени в пути RTG, в клетках 0 может быть обращена добавлением глутамата в среду. (15) Таким образом, глутамат подавляет путь RTG в дрожжах, хотя точные аминокислоты, ответственные за эту глутаматную репрессию генов-мишеней RTG, остаются неуловимыми, учитывая обширное взаимное превращение глутамата в глутамин и родственные аминокислоты. Комплекс SPS ( SSY1 , PTR3 , SSY5 ) представляет собой внешний аминокислотный сенсор в плазматической мембране. (16) SPS необходим для глутаматной репрессии генов пути RTG, а CIT2 ген конститутивно экспрессируется в SPS-дефицитных клетках (рис.). Knorre et al. (17) обсуждалось в недавнем обзоре, как дрожжи специфически обнаруживают митохондриальные функциональные нарушения. Как описано выше, разумно контролировать уровни глутамата как сигнал о митохондриальных нарушениях, поскольку глутамат, а также аргинин синтезируются в митохондриях. Однако путь RTG также активируется путем ингибирования мишени рапамицина (TOR), который является основным сенсором питательных веществ в цитоплазме. Следовательно, обнаружение измененных уровней аминокислот с помощью пути RTG может быть неспецифичным для митохондриальных нарушений.Другие митохондриальные продукты, такие как АТФ и железосернистый кластер, а также пептиды митохондриального происхождения, также могут использоваться для индикаторов митохондриальной функции. (17) Фактически низкая концентрация АТФ диссоциирует Rtg2 от Mks1 и активирует путь RTG (рис.). Однако снижение АТФ возможно без нарушения митохондрий при культивировании в среде с высоко ферментируемыми источниками углерода. Таким образом, реакции РИТЭГа, по всей видимости, нет.
специфичны для митохондриальных нарушений и зависят от различных клеточных контекстов и метаболического статуса.Напротив, система нематод ATFS-1 непосредственно отслеживает нарушение митохондрий, исследуя эффективность импорта митохондриального белка, которая зависит от потенциала митохондриальной мембраны (18) (также обсуждается позже). Системы, гомологичные ATFS-1, в настоящее время неизвестны в клетках дрожжей или млекопитающих, и те же вопросы специфичности могут также применяться к определенным путям MRS у млекопитающих.
). (13) По существу, ингибирование MRS в клетках ρ 0 делает дрожжевые ауксотрофы глутамата. CIT2 , ген, важный для пероксисомального глиоксилатного цикла, и несколько других генов-мишеней MRS контролируются Rtg1p, Rtg2p и Rtg3p (13,14) (далее называемый путем RTG). Rtg2p необходим для активации факторов транскрипции bHLH-Zip, Rtg1p и Rtg3p в клетках ρ 0 . Отмеченная повышающая регуляция гена CIT2 , канонического гена-мишени в пути RTG, в клетках 0 может быть обращена добавлением глутамата в среду. (15) Таким образом, глутамат подавляет путь RTG в дрожжах, хотя точные аминокислоты, ответственные за эту глутаматную репрессию генов-мишеней RTG, остаются неуловимыми, учитывая обширное взаимное превращение глутамата в глутамин и родственные аминокислоты. Комплекс SPS ( SSY1 , PTR3 , SSY5 ) представляет собой внешний аминокислотный сенсор в плазматической мембране. (16) SPS необходим для глутаматной репрессии генов пути RTG, а CIT2 ген конститутивно экспрессируется в SPS-дефицитных клетках (рис.). Knorre et al. (17) обсуждалось в недавнем обзоре, как дрожжи специфически обнаруживают митохондриальные функциональные нарушения. Как описано выше, разумно контролировать уровни глутамата как сигнал о митохондриальных нарушениях, поскольку глутамат, а также аргинин синтезируются в митохондриях. Однако путь RTG также активируется путем ингибирования мишени рапамицина (TOR), который является основным сенсором питательных веществ в цитоплазме. Следовательно, обнаружение измененных уровней аминокислот с помощью пути RTG может быть неспецифичным для митохондриальных нарушений.Другие митохондриальные продукты, такие как АТФ и железосернистый кластер, а также пептиды митохондриального происхождения, также могут использоваться для индикаторов митохондриальной функции. (17) Фактически низкая концентрация АТФ диссоциирует Rtg2 от Mks1 и активирует путь RTG (рис.). Однако снижение АТФ возможно без нарушения митохондрий при культивировании в среде с высоко ферментируемыми источниками углерода. Таким образом, реакции РИТЭГа, по всей видимости, нет.
специфичны для митохондриальных нарушений и зависят от различных клеточных контекстов и метаболического статуса.Напротив, система нематод ATFS-1 непосредственно отслеживает нарушение митохондрий, исследуя эффективность импорта митохондриального белка, которая зависит от потенциала митохондриальной мембраны (18) (также обсуждается позже). Системы, гомологичные ATFS-1, в настоящее время неизвестны в клетках дрожжей или млекопитающих, и те же вопросы специфичности могут также применяться к определенным путям MRS у млекопитающих.
Активация MRS изменяет метаболизм митохондрий у дрожжей. Глутаматное голодание и дыхательная недостаточность митохондрий в истощенных мтДНК клетках (ρ 0 клеток) активируют MRS для экспрессии генов, вовлеченных в митохондриальный цикл TCA ( CIT1 , ACO1 и IDh2 / 2 ) и цикл пероксисомального глиоксилата (). CIT2 ).Когда путь RTG не задействован, основные факторы транскрипции лейциновой молнии спираль-петля-спираль, Rtg1 и Rtg3, секвестрируются в цитоплазму, где Rtg3 гиперфосфорилируется. Mks1 способствует гиперфосфорилированию Rtg3, каким-то образом предотвращая дефосфорилирование Rtg3 и негативно регулируя этот путь. Когда включается ретроградный путь, Rtg3 становится частично дефосфорилированным, проникает в ядро вместе с Rtg1 и связывает R-бокс для активации транскрипции. Rtg2, цитоплазматический белок с N-концевым АТФ-связывающим доменом, располагается выше Rtg1 и Rtg3.В отсутствие Rtg2 CIT2 больше не реагирует на митохондриальные аномалии. Rtg2 связывается и инактивирует Mks1, обеспечивая активацию Rtg1 / 3 и пути RTG. Физиологические концентрации АТФ диссоциируют Mks1 от Rtg2 и негативно регулируют этот путь. (109) Комплекс SPS является сенсором для внешних аминокислот и важен для глутаматной репрессии пути RTG, а инактивация сенсорной системы SPS приводит к Rtg2-зависимому увеличению экспрессии CIT2 и потере репрессии глутамата. .Lst8, который является неотъемлемым компонентом мишеней комплексов рапамицина (TOR), также является негативным регулятором пути RTG и регулирует путь RTG в двух точках: выше и ниже Rtg2. Rtg1 / 3-зависимая экспрессия гена активируется в клетках, в которых передача сигналов TOR, опосредованная родственными киназой PI киназами, Tor1 и Tor2, ингибируется иммунодепрессантом рапамицином. Дисфункция митохондрий индуцирует альтернативный ретроградный путь передачи сигналов, чтобы индуцировать экспрессию ATO3 способом, зависящим от SPS и Gcn4, но не от Rtg2.ААР, аминокислотная пермеаза; ACO1, аконитатгидратаза; α-KG, α-кетоглутарат; ATO3, вынос аммиака наружу 3; CIT, цитрат-синтаза; IDH, изоцитратдегидрогеназа; Lst8, летальный с сек тринадцать 8; Mks1, мультикопийный супрессор киназы 1; ОАА, оксалоацетат; РИТЭГ, ретроградное регулирование; СПС, ССы1п-Птр3п-ССы5.
Помимо RTG-зависимых генов, ATO3 , ген, важный для экскреции аммония, также активируется в клетках ρ 0 . Gcn4, главный регулятор реакции аминокислотного голодания у дрожжей, вносит вклад как в базальную, так и в индуцируемую ρ 0 экспрессию ATO3 , а SPS-зависимый сигнал, в частности, способствует индуцируемой ρ 0 экспрессии ATO3. . (19) Gcn4 также активируется в дрожжах, лишенных митохондриального гена протеазы AAA AFG3 , и приводит к увеличению продолжительности жизни. (20)
Обзор ретроградной передачи сигналов митохондрий млекопитающих
Роль митохондрий и MRS может быть различной и разнообразной у разных видов, особенно между одноклеточными эукариотами и сложными многоклеточными организмами, такими как млекопитающие. У многоклеточных животных митохондрии необходимы не только для эмбрионального развития, но и для различных функций взрослых организмов, включая воспаление, иммунитет и апоптоз.У млекопитающих MRS часто обсуждается с точки зрения контроля качества и адаптивного биогенеза митохондрий. Например, нарушение митохондрий снижает выработку АТФ и активирует AMPK, который активирует PPARγ-коактиватор (PGC) 1α-опосредованный митохондриальный биогенез, чтобы противодействовать снижению АТФ (рис.). Митохондрии функционируют как внутриклеточные хранилища Ca 2+ не только для буферизации цитоплазматического Ca 2+ в покоящихся клетках, но также для стимуляции синтеза АТФ путем активации транспорта Ca 2+ и Ca 2+ -зависимых ферментов в цикле TCA и OxPhos в активированных миоцитах. (21) Снижение митохондриального дыхания приводит к потере митохондриального мембранного потенциала (MMP), что снижает приток митохондриального Ca 2+ и приводит к устойчивому увеличению цитоплазматического Ca 2+ и активации кальциневрина (Cn) и кальций / кальмодулин-зависимая киназа (CaMK). Cn активирует гликолиз и OxPhos путем экспрессии транспортера глюкозы 4 (Glut4) и субъединицы 5B цитохром-с-оксидазы (COX5B) через NF-κB и NFAT, соответственно. (22,23) CaMK активирует CREB, который индуцирует экспрессию цитохрома c, COX2, PGC1α и митохондриального транскрипционного фактора A (TFAM) для увеличения активности OxPhos и митохондриального биогенеза. (24) CaMK также активирует AMPK, который регулирует PGC1α и FOXO, что приводит к активации митохондриального биогенеза. Многие экологические стрессы, в том числе УФА, индуцируют митохондриальные АФК, которые активируют путь Nrf2. (25) Кроме того, сообщается, что введение окиси углерода активирует сердечный Nrf2 за счет генерации митохондриальных АФК. (26)
ATF4 действует как стресс-чувствительный фактор транскрипции
Активирующий фактор транскрипции 4 (ATF4) является основным транскрипционным фактором лейциновой молнии (bZip), принадлежащим к подсемейству ATF / CREB.ATF4 активируется различными стрессами, такими как стресс эндоплазматического ретикулума (ER) и дефицит аминокислот. (27) ATF4 образует гомодимеры или гетеродимеры с другими факторами транскрипции, такими как C / EBPβ, и функционирует как трансактиватор или репрессор. Гетеродимер ATF4 / C / EBPβ распознает консенсусную последовательность ДНК [аминокислотно-ответный элемент (AARE) или C / EBP-ATF-ответный элемент (CARE)], консенсусной последовательностью которой является TGATGNAAN. Гетеродимер ATF4 / C / EBPβ активирует экспрессию нижележащих генов-мишеней, таких как аспарагинсинтетаза ( ASNS ) и гомолог 3 трибблз ( Trib3 ).ATF4 модулирует многие важные биологические процессы, такие как антиоксидантный ответ, апоптоз, энергетический гомеостаз, аутофагия и остеогенез. Некоторые аномалии, включая остеопороз, микрофтальм и аномальный гематопоэз, обнаруженные у мышей с нокаутом ATF4 (KO), отражают значительную роль ATF4 в развитии млекопитающих. Активация ATF4 стрессовыми сигналами опосредуется консервативным сигнальным путем, называемым интегрированным стрессовым ответом (ISR). Фосфорилирование Ser51 фактора инициации эукариот (eIF) 2α различными киназами eIF2α приводит к селективной трансляции ATF4 и глобальному ингибированию трансляции. (28) В клетках млекопитающих четыре киназы eIF2α, чувствительные к стрессу, регулируемый гемом ингибитор трансляции (HRI), активируемая дцРНК протеинкиназа (PKR), PKR-подобная киназа эндоплазматического ретикулума (PERK) и общий контроль недерепрессируемый-2 ( GCN2), катализируют фосфорилирование eIF2α в ответ на соответствующие стрессовые сигналы. Каждая киназа eIF2α активируется различными стрессовыми сигналами, например HRI (дефицит гема), PKR (вирусная инфекция), PERK (стресс ER) и GCN2 (дефицит аминокислот). Примечательно, что фосфорилирование eIF2α снижает связанный с GTP eIF2 (GTP-eIF2) за счет секвестрации eIF2α.Когда GTP-eIF2 в изобилии, глобальная трансляция поддерживается, в то время как трансляция ATF4 ингибируется из-за двух вышестоящих открытых рамок считывания (uORF) в 5′-нетранслируемой области (5′-UTR) мРНК ATF4. Напротив, глобальная трансляция замедляется при низком уровне GTP-eIF2, но низкий уровень GTP-eIF2 позволяет рибосомам обходить ингибирующую uORF и активировать селективную трансляцию ATF4.
Активация ATF4 при митохондриальных заболеваниях
Митохондриальная дисфункция, вызванная мутациями в митохондриальной ДНК (мтДНК), связана с множеством заболеваний, включая митохондриальную миопатию, энцефалопатию, молочнокислый ацидоз, приступы инсульта и ретрогеницита (MELAS) и нейрогенициты. Пигментные (НАРП) синдромы. (2) Чтобы повторить клеточный ответ на мутации митохондриальных генов, Fujita et al. (29) сгенерировал человеческие гибридные клетки остеосаркомы 143B, несущие митохондрии с мутацией типа MELAS (мутация 3243A> G в гене митохондриальной тРНК для лейцина), называемые клетками 2SD, и мутацию типа NARP (мутация 8993T> G, приводящая к замена аминокислоты с высококонсервативного лейцина в положении 156 на аргинин в субъединице 6 митохондриальной F o F 1 -АТФаза), называемых клетками NARP3-1.Авторы показали, что ATF4 активируется в этих клетках, что сопровождается индукцией его целевого белка CHOP и ASNS. (29) Эти авторы также показали, что индукция CHOP и ASNS также наблюдается ротеноном, ингибитором комплекса I mETC. Cortopassi et al. (30) систематически исследовал клеточный ответ на митохондриальные мутации с использованием по крайней мере 2 репрезентативных типов клеток пяти митохондриальных заболеваний, включая наследственную оптическую невропатию Лебера (LHON), атаксию Фридрейха (FRDA), NARP, миоклоническую эпилепсию с рваными красными волокнами. (MERRF) и синдром Кирнса – Сейра (KSS).Лучшая категория индуцибельных генов была связана с пролиферацией и клеточным циклом, а также с развернутой стрессовой реакцией, которая регулируется ATF4.
Активация ATF4 функциональным нарушением митохондрий
Накапливающиеся данные продемонстрировали, что несколько различных стимулов, нарушающих функции митохондрий, активируют путь ATF4 (рис. И таблица).
ATF4 активируется при нарушении функции митохондрий. Нарушение функции митохондрий вызывает активацию ATF4.Генетическое изменение или ингибирование ферментов OxPhos увеличивает продукцию ROS, что может вызвать активацию ATF4. ATF4 также активируется нарушением регуляции других метаболических путей, таких как накопление реактивного альдегида за счет доминирующей отрицательной экспрессии ALDh3 или нарушение гомеостаза железа из-за дефицита FXN. Митофагия регулирует контроль качества митохондрий, удаляя поврежденные деполяризованные митохондрии. Нарушения митофагии вызывают накопление поврежденных митохондрий и активацию ATF4.Дисбаланс митохондриальных и ядерно-кодируемых митохондриальных белков, вызванный делецией мтДНК или ингибированием митохондриальных рибосом доксициклином, вызывает активацию ATF4. FXN, фратаксин; OxPhos, окислительное фосфорилирование.
Таблица 1
In vivo свидетельства активации ATF4 митохондриальным стрессом
| Ткань | Доказательства | Ref | Другие | ||||||
|---|---|---|---|---|---|---|---|---|---|
| мутантная мутация мутантной системы трансгенных мутантов геликаза (мыши deletor) | Мышцы | Индукция генов Mthfd2 и Fgf21 | (29) | Ассоциирует с активацией AKT | |||||
| Мыши с дефицитом Cox10 | Мышцы | Mthf2175 и индукция гена Fgf275 Ассоциирует с активацией AKT | |||||||
| Лечение метформином | Печень | Повышенная индукция белка ATF4 и гена Fgf21 | (30) | ||||||
| Скелетные мышцы, специфичные для UCP1, гиперэкспрессия Fgf2 | индукция гена 907 52 | (32) | |||||||
| Системная экспрессия доминантно-отрицательного ALDh3 | Сердце | Повышенная индукция целевого гена ATF4 | (36) | Гетерозиготная делеция ATF4 уменьшала ответ | 2 | F на мускулатуру | Повышенная индукция гена-мишени ATF4 | (37) | |
| Нокаут ATG7, специфичный для скелетных мышц | Скелетная мышца | Повышенная индукция гена Fgf21 в скелетных мышцах | -специфический двойной нокаут Mfn1 и Mfn2 | Скелетная мышца | Повышенное фосфорилирование белка ATF4 и eIF2α в скелетных мышцах | (38) |
Активация ATF4 посредством
прогрессирующего внешнего ингибирования OxiaPhos2 ) является прогрессирующим мЭТК с началом у взрослых дефицит с множественными делециями мтДНК. (31,32) Мыши Deletor представляют собой модели PEO, в которых доминантная мутация мтДНК геликазы Twinkle приводит к делеции мтДНК и позднему началу дефицита мЕТС. (33) У мышей deletor, а также мышей с дефицитом фактора сборки цитохрома c 10 (Cox10) в мышцах индуцируются гены-мишени ATF4, включая фактор роста фибробластов (FGF) 21 и Mthfd2. (34) AKT1-фосфорилирование треонина 308 и серина 473 активируется в скелетных мышцах мышей deletor, что указывает на участие AKT1 в активации ATF4, вызванной митохондриальным стрессом.Эти авторы далее показали, что избыточная экспрессия PGC1α улучшает дефицит mETC и снижает индукцию FGF21 и Mthfd2 у мышей с дефицитом Cox10. Метформин, агент, снижающий уровень глюкозы, клинически используемый для лечения диабета, ингибирует mETC. Kim et al. (35) показали, что метформин индуцирует экспрессию гена FGF21 и уровни белка ATF4 в печени мышей. Метформин подавляет активность комплекса I в клетках FaO гепатомы крысы и индуцирует экспрессию гена FGF21 посредством активации ATF4 в клетках FaO и эмбриональных фибробластах мыши (MEF). (35) Кроме того, митохондриальный антиоксидант Mito-TEMPO, обладающий активностью улавливания супероксидных и алкильных радикалов, ингибирует индуцированную метформином активацию ATF4 в клетках FaO, что указывает на роль митохондриальных АФК. Кроме того, индуцированная метформином активация ATF4 является PERK-зависимой, что указывает на то, что метформин индуцирует ATF4 в митохондриальном пути ROS-PERK (обсуждается позже). Избыточная экспрессия разобщающего белка 1 (UCP1) в скелетных мышцах улучшает инсулинорезистентность и продлевает продолжительность жизни мышей, получавших диету с высоким содержанием жиров. (36) Кроме того, Keipert et al. (37) продемонстрировали, что сверхэкспрессия UCP1 в скелетных мышцах активирует экспрессию генов ATF4 и FGF21. Эти авт. Показали, что как избыточная экспрессия UCP1, так и ингибирование митохондриального дыхания потенциально снижают потенциал митохондриальной мембраны, активируют ATF4 в мышечных трубках C2C12. LHON вызывается мутацией субъединиц комплекса I. Подражая LHON, Silva et al. (38) продемонстрировали, что ингибитор комплекса I, ротенон, индуцирует ATF4, который связан с активацией PERK в олигодендроцитах человека.Ротенон также индуцировал PERK-зависимую активацию JNK в этих клетках. В клетках HCT116 ингибирование АТФ-синтазы олигомицином, которое вызывает гиперполяризацию митохондрий, в отличие от ингибитора mETC, вызывает перепрограммирование энергетического метаболизма, при котором функции митохондрий подавляются. (39) Индуцированные олигомицином AMPK и ATF4 могут быть ответственны за репрессию митохондрий. Ингибирование комплекса I и III mETC индуцирует экспрессию SESN2 в линиях раковых клеток ATF4-зависимым образом. (40)Активация ATF4 посредством метаболической дисфункции
Новаторская работа Endo et al. (41) продемонстрировали, что накопление реактивных альдегидов из-за экспрессии доминирующего отрицательного типа митохондриальной альдегиддегидрогеназы 2 (ALDh3) активирует ATF4 в сердце и провоцирует цитопротекторный ответ против ишемического реперфузионного повреждения сердца. FRDA вызывается мутациями в гене FXN, который кодирует белок митохондриального матрикса фратаксин, который необходим для биосинтеза кластера железа и серы в митохондриях.У мышей с мышечно-специфическим нокаутом FXN ATF4 активируется с помощью до сих пор неизвестных механизмов, предшествующих функциональному и анатомическому ухудшению состояния сердца. (42)
Активация ATF4 дефектами митофагии
Митофагия — важная система контроля качества поврежденных или функционально дефицитных митохондрий. У мышей KO, специфичных для мышц ATG7, функция митохондрий, оцениваемая по состоянию 3 и состоянию 4 дыхания, была снижена, а экспрессия гена FGF21 была активирована через ATF4. (43) Паркин и PINK1 мутированы при семейных типах болезни Паркинсона и участвуют в контроле качества митохондрий. В Drosophila ATF4 индуцируется в головах мутантов pink1 и parkin, которые являются хорошо известными моделями болезни Паркинсона у Drosophila и нарушают функцию митохондрий. (44)
Активация ATF4 дефектами митохондриальной динамики
Митохондриальная динамика слияния и деления тесно связана с функцией митохондрий.Более того, специфическая делеция митофузина 1 (Mfn1) и 2 (Mfn2) также активирует передачу сигналов ATF4 в мышцах. (43) Этот эффект, скорее всего, опосредован активацией Perk, потому что нокаут Mfn2 вызывает стресс ER и активирует Perk. (45) Соответственно, нокаут атрофии зрительного нерва 1 (OPA1) также вызывает стресс ER и FGF21. (46)
Активация ATF4 мито-ядерным дисбалансом
Дисбаланс митоядерных белков может быть вызван различными механизмами, включая мутацию мтДНК, ингибирование трансляции митохондрий или репрессию ядерных генов, и может способствовать долголетию многих видов. (47) Ингибитор митохондриальной трансляции доксициклин (DOX) и другие тетрациклины также вызывают активацию ATF4 в различных раковых клетках. (48,49) Бао и др. (50) продемонстрировали, что истощение митохондриальной ДНК, индуцированное экспрессией доминантно-отрицательного мутанта ДНК-полимеразы γ или бромида этидия, активирует ATF4 в клетках HEK293.
UPR млекопитающих
MT и ATF4UPR возникает, когда количество неправильно свернутых белков превышает способность клетки к укладке белков.Первоначально UPR был описан в отношении ER. Однако недавно появилось сообщение об UPR митохондрий. (6) Этот путь был впервые описан у млекопитающих, у которых митохондриальные шапероны индуцируются делецией митохондриальной ДНК, (51) , но после этого большинство механизмов было выяснено у C. elegans . Митохондрии обладают собственными шаперонами и протеазами, а экспрессия их генов активируется UPR MT . В C. elegans развернутые белки расщепляются митохондриальной протеазой Clp, и полученные пептиды транспортируются внутренним транспортером митохондриальных пептидов HAF-1 и впоследствии диффундируют в цитоплазму.Фактор транскрипции ATFS-1 обычно перемещается в митохондрии и разрушается протеазой LON, но снижение митохондриального импорта ATFS-1 приводит к ядерной транслокации ATFS-1 и активирует UPR MT . И снижение импорта митохондриального белка, и ядерная транслокация ATFS-1 зависят от оттока HAF-1 и пептидов; однако точный механизм и жесткое регулирование остаются неясными. Хотя ATFS-1 и HAF-1 не существуют у млекопитающих, новые данные показали, что функционально гомологичная система также существует у млекопитающих.Munch et al. (52) показали, что ингибиторы митохондриального шаперонина, HSP90 / TRAP1 GTPP и LON протеазы CDDO, приводят к глобальным изменениям в экспрессии генов, которые связаны с дефектами процессинга митохондриальной пре-РНК, что приводит к ингибированию синтеза митохондриального белка. Гены, активируемые острым стимулом UPR MT млекопитающих, включают митохондриальные шаперонины HSPD1 и HSPE1, а также несколько генов-мишеней ATF4, таких как CHOP. Промоторы генов, индуцированные как GTPP, так и CDDO, показали обогащение по сайтам связывания для CHOP и ATF4, а также митохондриального UPR-ответного элемента (MURE1 и MURE2), подтверждая роль ATF4 в UPR MT млекопитающих.ATF4 может быть активирован мито-ядерным дисбалансом, вызванным ингибированием трансляции митохондрий. Однако эти авторы показали, что нокдаун любой из четырех киназ eIF2α не влияет на индукцию гена CHOP с помощью GTPP, указывая на независимый от фосфорилирования eIF2α механизм активации ATF4 с помощью GTPP.
Киназы EIF2α и АФК как кандидаты в вышестоящие регуляторы активации ATF4 митохондриальным возмущением
Bao et al. (50) показали, что ингибиторы OxPhos, такие как антимицин и олигомицин, но не митохондриальный протонофор CCCP, сильно активируют ATF4 в клетках HEK293, что указывает на то, что активация ATF4, вызванная ингибитором OxPhos, включает окислительно-восстановительный стресс, но не истощение АТФ и потерю MMP. .Bouman et al. (53) показали, что митохондриальная дисфункция, запускаемая CCCP, вызывает стресс ER и активирует индукцию ATF4-зависимого гена паркина через путь UPR ER -PERK в клетках SH-SY5Y. (53) Интересно, что паркин также индуцируется туникамицином или тапсигаргином и предотвращает снижение АТФ-синтазы и жизнеспособности клеток, предотвращая фрагментацию митохондрий. Таким образом, паркин защищает митохондриальные нарушения, вызванные стрессом ER.Более того, метформин индуцирует ATF4 AMPK-независимым, но зависимым от PERK образом. (35) Становится все более очевидным, что ассоциированная с митохондриями мембрана (MAM) играет ключевую роль в клеточной передаче сигналов и коммуникации между митохондриями и ER. (54) Интересно, что CO активирует ATF4 через PERK митохондриальным ROS-зависимым образом, указывая на то, что митохондриальные нарушения индуцируют ATF4 через связь MAM. (55) Однако некоторые данные предполагают участие GCN2 в качестве вышестоящего регулятора активации ATF4, о чем сообщалось у нематод. (56) Мартинес-Рейес et al. (39) обнаружили, что GCN2 активируется олигомицином, и постулировали, что путь GCN2-ATF4, а также AMPK, отвечает за метаболическую адаптацию (то есть репрессию митохондрий) к олигомицину в клетках рака толстой кишки HCT116. (39) Повышенное фосфорилирование eIF2α под действием DOX ингибировалось нокдауном GCN2 в клетках HeLa. (48) Ван и др. (57) наблюдали, что митохондриальная дисфункция в клетках рака желудка увеличивает устойчивость к цисплатину за счет индукции транспортера цистеина xCT. (57) Эти авторы показали, что активация ATF4, вызванная митохондриальной дисфункцией, зависит от GCN2, но не от PERK. Следовательно, определенные митохондриальные токсические стимулы, такие как CCCP и ингибитор комплекса I, вероятно, вызывают стресс ER и индуцируют ATF4 PERK-зависимым образом, в то время как другие токсические стимулы против митохондрий индуцируют ATF4 в зависимости от GCN2 и клеточного типа.
В состоянии дефицита глюкозы анаплеротический путь от пирувата снижается, что приводит к снижению метаболитов цикла TCA.Затем глутамат используется для обеспечения α-кетоглутарата для восполнения цикла TCA (т. Е. Анаплеротического пути), что приводит к недостаточности глутамата. (58) Действительно, путь GCN2-ATF4 индуцируется дефицитом глюкозы, связанным с уменьшением количества промежуточных продуктов цикла TCA и нескольких аминокислот. (59) В пролиферирующих клетках ингибирование ETC приводит к дефициту аспартата из-за недостаточности оксалоацетата. (60) Следовательно, как и в случае с дрожжами, аминокислотное голодание также может быть сигналом для MRS у млекопитающих, хотя оно сильно зависит от доступности аминокислот из окружающей среды.Эту гипотезу следует изучить в будущих анализах. Учитывая, что нематодный путь GCN-2-ATF4 также активируется в ответ на митохондриальные пертурбации, (56) путь GCN-2-ATF4 может быть эволюционно консервативным путем, который отвечает на митохондриальные пертурбации, ощущая активность цикла TCA. .
Механизмы ATF4-опосредованного контроля качества митохондрий
ATF4 регулирует гены, связанные с внутриклеточным гомеостазом аминокислот, такие как переносчик аминокислот и синтаза аминокислот, а также гены антиоксидантов, такие как xCT. (27) Кроме того, ATF4 регулирует гены, связанные с апоптозом, такие как CHOP, и усиливает апоптоз, когда стресс ER является тяжелым или продолжительным. (61) Однако то, как ATF4 регулирует функцию митохондрий, изучено лишь частично. Если ATF4 активируется митохондриальными стимулами и защищает от митохондриальной дисфункции, то каковы основные механизмы? Несколько генов, важных для митохондриальных функций, регулируются ATF4.
Паркин
Путь PINK1-Parkin играет важную роль в контроле качества митохондрий, обнаруживая повреждение митохондрий и, как следствие, индуцируя митофагию поврежденных митохондрий. (62) Как упоминалось выше, Bouman et al. (53) показали, что ATF4 регулирует паркин, способствуя контролю качества митохондрий. Эти авторы также показали, что ATF4 связывается с сайтами CREB / ATF4 гена Паркина и что c-Jun негативно регулирует экспрессию гена Паркина в той же области.
Митохондриальный фолат-опосредованный метаболизм одной углеродной единицы (1С), синтез серина и путь транссульфурации
Фолат-опосредованный метаболизм 1С, а также путь синтеза и транссульфурации серина, а также путь метилирования являются высоко взаимосвязанными метаболическими путями. (63) Недавний анализ показал, что большая часть серина метаболизируется в митохондриях, а выработка митохондриального формиата имеет решающее значение для цитозольной продукции пуринов и тимидинов в большинстве линий раковых клеток. (64) ATF4 недавно был признан центральным игроком, регулирующим экспрессию гена митохондриального метаболизма 1С, а также генов синтеза серина и пути транссульфурации, как показано вышеупомянутыми результатами (рис.). (41,65) Учитывая дегенеративные заболевания головного мозга, синтез серина de novo (т.е.е., путь фосфорилирования серина) важен, потому что гематоэнцефалический барьер не переносит в достаточной степени серин с пищей. (66,67) У мышей с моделью митохондриального заболевания эти пути активированы, что приводит к ремоделированию одноуглеродного метаболизма. (68) Кроме того, ATF4 регулирует эти пути, способствуя прогрессированию рака, способствуя биосинтезу пуринов. (69) Важно отметить, что митохондриальный метаболизм 1С также важен для митохондриального синтеза побочных продуктов НАДФН, необходимых кофакторов для биосинтеза антиоксидантов и липидов в митохондриях. (70,71) У описанных выше мышей, экспрессирующих доминантно-отрицательный ALDh3, заметно индуцируется экспрессия генов Mthfd2 и Phgdh, которые являются ферментами, ограничивающими скорость митохондриального метаболизма 1С и пути синтеза серина, соответственно. (41) Эти авторы наблюдали увеличение клеточного GSH, хотя митохондриальная генерация NADPH, вероятно, также увеличивается, что вносит вклад в фенотип устойчивости к окислительному стрессу у мышей. У мух с мутантами parkin и pink1 экспрессия генов белков Shmt2, Mthfd2, GCS P и T, которые участвуют в митохондриальном фолат-опосредованном метаболизме 1C, генерирующем продукцию митохондриального NADPH, повышается за счет активации ATF4. (44) По-видимому, активация метаболизма 1С в данном случае не приводит к пролиферации клеток, но, вероятно, активирует продукцию НАДФН в митохондриях. Более того, Shmt2 важен для de novo синтеза dTTP и репликации митохондриальной ДНК, (72) , а также для трансляции кодируемой митохондриями ETC посредством регуляции метилирования тРНК, как недавно сообщалось у млекопитающих. (73) Более того, активация ATF4 в сердце сверхэкспрессией каталитической субъединицы β1 кальциневрина (CnAβ1) (обсуждается позже) защищает митохондриальное окисление белков и оказывает защитную функцию против опосредованной перегрузкой сердца гипертрофии сердца в зависимости от PHGDH и глутатиона. (74) Следовательно, митохондриальный метаболизм 1С, регулируемый ATF4, может быть важен для митохондриального биогенеза и митохондриальной антиоксидантной функции (рис.).
ATF4 регулирует синтетический серин и митохондриальный путь метаболизма 1С, опосредованный фолатом. Несколько генов-мишеней ATF4 участвуют в синтезе серина, метаболизме 1С единиц, синтезе пурина и синтезе глутатиона. Экспрессия PHGDH, PSAT1 и PSPH увеличивает синтез серина. Серин участвует в синтезе глутатиона, а также в метаболизме единиц 1С.Экспрессия xCT, CTH и CBS увеличивает синтез глутатиона. Экспрессия SHMT2 и MTHFD2 ускоряет продукцию NADPH, синтез fMet и синтез пуринов, которые важны для митохондриального окислительно-восстановительного гомеостаза, митохондриальной трансляции и репликации ДНК, соответственно. SHMT2-опосредованное метилирование тРНК важно для трансляции кодируемой митохондриями транспортной цепи электрофилов. В пролиферирующих клетках CHO-THF и Ch3-THF, образующиеся в цитозоле, используются для синтеза пурина и тимидина соответственно.3ПГ, 3-фосфоглицерат; 3PHP, 3-фосфогидроксипируват; CBS, цистатионин-β-синтаза; CH 2 -THF, 5,10-метилен-THF; CH + -THF, 5,10-метенил-THF; СНО-ТГФ, 10-формил-ТГФ; CTH, цистатионаза; dTMP, монофосфат тимидина; дТТФ, тимидинтрифосфат; fMet, N -формилметионин; MTHFD2, метилентетрагидрофолатдегидрогеназа 2; PHGDH, фосфоглицератдегидрогеназа; PSAT1, фосфосерин-аминотрансфераза 1; PSer, 3-фосфосерин; PSPH, фосфосеринфосфатаза; SHMT2, митохондриальная серингидроксиметилтрансфераза 2; ТГФ, тетрагидрофолат.
FGF21
ATF4 индуцировал миокин FGF21, как показано вышеупомянутыми результатами. (34,43) FGF21 обладает понижающим уровень глюкозы и антилипидемическим действием у тучных животных, и этот белок может иметь хорошее влияние на метаболический синдром и долголетие. FGF21 регулируется Akt в скелетных мышцах и считается гормоном стресса. (75) Lehtonen et al. (76) утверждают, что FGF21 в значительной степени индуцируется дефектом митохондриальной трансляции или делецией мтДНК, но не так явно дефектом mETC.Это в некоторой степени согласуется с отчетом Dogan et al. (77) , что дефект митохондриальной аспартил-тРНК вызывает функциональную аномалию mETC как в сердце, так и в скелетных мышцах, но индукция FGF21 наблюдается только в сердце, утверждая, что индукция FGF21 не вызвана дефицитом mETC. , но нарушение протеостаза в митохондриях. Таким образом, как митохондриальные функциональные дефекты индуцируют экспрессию гена FGF21, еще предстоит выяснить в будущем.Эффекторный механизм FGF21 в значительной степени неизвестен, но этот белок улучшает функцию митохондрий посредством пути AMPK-SIRT1-PGC1α. (78)
Другие
ATF4 передает сигналы, способствующие выживанию, в условиях окислительного стресса, регулируя экспрессию гена MKP-1 и ингибируя проапоптотическую передачу сигналов MAPK. (79)
Активация пути ATF4 внеклеточными сигналами роста
В последнее время становится все более очевидным, что сигналы роста активируют путь ATF4, особенно в раковых клетках (рис.). Принимая во внимание, что путь ATF4 регулирует реакции аминокислотного голодания, включая индукцию переносчиков аминокислот на клеточной поверхности, важных для питания и роста клеток, разумно, что внешние сигналы роста также используют путь, опосредованный ATF4, для роста клеток. (80) В клеточной линии гиппокампа HT22 ингибитор PI3K LY294002 снижал фосфорилирование eIF2α, что было связано со снижением экспрессии xCT и ATF4. Кроме того, ингибитор GSK3β, хлорид лития, восстанавливает вызванное ингибированием PI3K снижение ATF4 GCN2-зависимым образом, указывая на то, что путь PI3K-Akt-GSK3β-GCN2-eIF2α участвует в базальной экспрессии ATF4 (рис.). В соответствии с этими наблюдениями, инсулин надежно активирует ATF4 в HT22 (81) и умеренно активирует ATF4 в MEF. (69) Интересно, что mTORC1, другая мишень Akt, активирует ATF4, чтобы впоследствии активировать синтез пурина в раковых клетках. (69) Кроме того, нейрон-специфическая делеция TSC2, отрицательного ингибитора mTORC1, активирует передачу сигналов mTORC1 и увеличивает экспрессию генов ATF4 и MTHFD2 в головном мозге. (69) Индукция ATF4 туникамицином была отменена в MEF с нокаутом мутанта Ser51Ala eIF2α [eIF2α (A / A) MEF].Однако индуцированное инсулином накопление ATF4 ингибировалось ингибитором mTORC1 рапамицином, но только частично уменьшалось в MEF eIF2α (A / A) по сравнению с MEF дикого типа, что указывает на неканонический механизм mTOR-опосредованной активации ATF4. (69) Недавно Park et al. (82) также сообщил, что mTORC1 активирует ATF4, регулируя eIF4E-связывающий белок (4E-BP). Более того, избыточная экспрессия Akt индуцирует экспрессию FGF21, предполагая, что Akt каким-то образом активирует ATF4 in vivo . (83) Примечательно, что mTORC1 регулирует митохондриальное дыхание, (84) и Akt локализуется в митохондриях после стимуляции PI3K, (85) , что указывает на возможное участие митохондрий в этих реакциях (рис.). Интересно, что недавнее исследование показало, что активация ATF4 выполняется за счет активации mTOR в пораженных мышечных волокнах при митохондриальной миопатии, предполагая, что mTOR локализуется выше по течению при активации ATF4 в ответ не только на внешние сигналы роста, но и на митохондриальные нарушения. (86) Однако CnAβ1, конститутивно активная форма каталитической субъединицы кальциневрина, лишенная C-концевого аутоингибиторного домена, также активирует ATF4 через путь mTORC2-AKT-mTORC1 в сердце, а сверхэкспрессия CnAβ1 улучшает сердечную функцию после сердечного приступа. инфаркт (87) или гипертрофия сердца, опосредованная перегрузкой давлением. (74) Интересно, что CnAβ1 индуцируется IGF-1 в сердечном сердце. (88)
Сигнал роста активирует ATF4 и метаболизм митохондрий.Активация ATF4 индуцируется не только сигналами стресса, которые вызывают фосфорилирование eIF2α и ингибирование трансляции, но также сигналами роста, которые активируют mTORC1 и ускоряют трансляцию. Сигналы роста активируют AKT, который фосфорилирует ряд субстратов в цитоплазме и митохондриях. Фосфорилирование TSC1 / 2 активирует mTORC1, который, в свою очередь, фосфорилирует S6K и 4E-BP для ускорения трансляции. Примечательно, что активация mTORC1 индуцирует экспрессию ATF4 без фосфорилирования eIF2α, но, вероятно, за счет истощения факторов повторной инициации трансляции.ATF4 индуцирует экспрессию гена в синтезе / транспорте аминокислот и синтезе аминоацил-тРНК в ответ на потребность в синтезе белка. 4E-BP, белок, связывающий фактор инициации эукариот 4E; ТСК, туберозный склероз.
Механизм активации AKT митохондриальным стрессом
Как описано выше, митохондриальный стресс активирует AKT, но его механизмы полностью не изучены. Истощение митохондрий или CCCP индуцировало экспрессию как инсулина, так и рецептора IGF-1 (IGF-1R), но избирательно активировало IGF-1R зависимым от Cn образом в линии миобластных клеток C2C12, которая отвечает за активацию Akt, приводящую к усиленному поглощению глюкозы. и инвазия опухоли. (89) Интересно, что эта индукция IGF-1R митохондриальным стрессом наблюдалась во многих линиях раковых клеток, включая A549, кардиомиоциты H9C2 и клетки NIh4T3. Мурата и др. (90) сообщил, что сверхэкспрессия PINK1 индуцирует фосфорилирование серина 473 AKT через mTORC2, предполагая, что митохондриальный стресс активирует AKT через активацию PINK1. Однако новаторская работа Kazemi et al. (91) продемонстрировали, что активация фосфорилирования eIF2α приводит к активации PI3K.Этот эффект может быть связан с подавлением негативной обратной связи регуляции PI3K через mTORC1. Они продемонстрировали, что фосфорилирование eIF2α подавляет mTORC1, а дерепрессированный PI3K активирует mTORC2-зависимое фосфорилирование AKT ATF4-независимым образом. (92) Таким образом, интерфероны и тапсигаргин индуцируют фосфорилирование S6 частично PKR- и PERK-зависимым образом, соответственно, в MEF. (91) Следовательно, фосфорилирование eIF2α может каким-то образом активировать PI3K в ответ на митохондриальные нарушения.
Активация ATF4 как самое раннее изменение при дегенеративных заболеваниях и токсических раздражителях — роль пути ISR-ATF4 для выживания
Длительная активация ATF4 часто приводит к апоптозу и предлагается для ускорения дегенеративных заболеваний, (61,93) , но несколько отчетов показали, что основная функция ATF4, по крайней мере, на ранней стадии стресса, является цитопротекторной. Как отмечалось выше, Huang et al. (42) наблюдали, что ранняя активация ATF4 предшествует началу ремоделирования сердца с использованием мышечно-специфичных мышей FXN KO, что указывает на то, что ATF4 действует как цитопротекторный фактор.Круг и др. (65) провел комбинированный анализ метаболомики и транскриптома в постмитотических дофаминергических нейронах человека и показал, что ATF4 активируется на ранней стадии (т.е. через 24 часа) после обработки митохондриальным токсикантом MPP + задолго до появления признака токсичности не наблюдалось (т. е. через 48 ч). Евстафьева и др. (94) исследовали роль пути ISR-ATF4 в защите от апоптоза в раковых клетках.Ингибитор митохондриального комплекса III миксотиазол активирует путь ATF4 в клетках HCT116 с ранней фазы временного цикла (~ 5 ч). Длительное ингибирование комплекса III, который специфически ингибирует синтез пиримидина de novo , активирует опосредованное p53 ингибирование экспрессии гена ATF4, что приводит к гибели клеток в результате апоптоза. Авторы утверждали, что ATF4 действует как защитный фактор против апоптоза, индуцированного митохондриальными ингибиторами. Таким образом, эти результаты показали, что ATF4 активируется митохондриальным стрессом на ранней, предсимптоматической стадии прогрессирования заболевания и защищает от последующего повреждения.Мы предполагаем, что активация ATF4 является биомаркером митохондриального стресса, который обещает вмешательство.
Профилактика возрастных или митохондриальных заболеваний с помощью активации Nrf2
Роль Nrf2 в контроле качества митохондрий
Nrf2 регулирует экспрессию сотен цитопротекторных генов для противодействия окислительному стрессу, вызванному эндогенно или экзогенно. (95) Эти генные продукты в основном уменьшают окислительный стресс в цитозоле, регулируя цитозольное количество глутатиона и НАДФН.Однако недавние исследования начинают раскрывать важную роль Nrf2 в контроле качества митохондрий и биогенезе, регулируя экспрессию NRF-1, PINK1 и TFAM (рис.). (96–99) Эффект Nrf2 в контроле качества митохондрий и биогенезе особенно очевиден в стрессовых условиях, таких как окислительный стресс или бактериальная инфекция, когда не происходит активации вышеупомянутых генов в нормальных гомеостатических условиях, таких как Нокаут-клетки Keap1. Nrf2 также специфически активируется в сердце мышей surf1 — / — с дефицитом Complex IV, что позволяет предположить, что Nrf2 играет роль в митохондриальных ретроградных ответах тканеспецифическим образом. (100)
Кооперативная регуляция генов для контроля качества митохондрий с помощью Nrf2 и ATF4. Становится ясно, что митохондриальный стресс активирует ATF4. В некоторых тканях, например в сердце, митохондриальные стрессы также могут активировать Nrf2 тканезависимым образом. ATF4 и Nrf2 индуцируют определенные гены стрессовой реакции (шапероны ER и синтетические гены аминокислот с помощью ATF4 и гены антиоксидантов с помощью Nrf2), но оба фактора регулируют гены, которые важны для контроля качества митохондрий.Примечательно, что Nrf2 и ATF4 физически взаимодействуют. Nrf2 ингибирует связывание ATF4 с промоторной областью CHOP и кооперативно регулирует антиоксидантные гены, такие как HO-1 и xCT, а также антиапоптотический ген ORP150.
Использование перекрестных помех между Nrf2 и ATF4 в профилактике дегенеративных заболеваний
Ранее мы идентифицировали ATF4 как фактор, взаимодействующий с Nrf2 в дрожжевой двугибридной системе, используя Nrf2-C-концевую область (включая Neh6, Neh2 и Neh4) в качестве приманки (данные не показаны).He et al. (101) продемонстрировали, что Nrf2 и ATF4 кооперативно индуцируют экспрессию гемоксигеназы 1 (HO-1), показывая, что Nrf2 и ATF4 связываются с элементом стрессовой реакции в энхансере HO-1, хотя и с более низкой аффинностью связывания по сравнению с таковой у гетеродимер Nrf2-small Maf в анализе EMSA с использованием рекомбинантных белков. Мы также протестировали связывание гетеродимера Nrf2-ATF4 с элементом антиоксидантного ответа в анализе EMSA с использованием рекомбинантных белков, но мы не обнаружили какого-либо значительного связывания.Вместо этого мы показали, что Nrf2 и ATF4 кооперативно активируют экспрессию гена xCT, субъединицы цистеинового транспортера xc —, в клеточных линиях рака мочевого пузыря. Nrf2 и ATF4 независимо связываются с сайтами связывания Nrf2 (ARE) и ATF4 (AARE) в регуляторной области и совместно регулируют транскрипцию гена xCT посредством своего физического взаимодействия. (102) Кроме того, Nrf2 и ATF4 совместно индуцируют другие антиоксидантные ферменты, такие как субъединицы γ-глутамилцистеинсинтазы и глутатионредуктаза, в ответ на антиоксидант карнозиную кислоту (Mimura J et al., 2018, неопубликованное наблюдение). Zong et al. (103) исследовали чувствительность клеток рака щитовидной железы к ингибиторам протеасом и показали, что Nrf2 ингибирует апоптоз, отрицательно влияя на положительный эффект ATF4 на экспрессию CHOP. (103) Эти авторы продемонстрировали, что Nrf2 влияет на связывание ATF4 с регуляторными областями CHOP. Более того, Nrf2 и ATF4 кооперативно регулируют экспрессию антиапоптотического молекулярного шаперона ORP150, локализованного как в ER, так и в митохондриях. (104) Таким образом, эти результаты продемонстрировали, что Nrf2 кооперативно индуцирует антиоксидантные ферменты и антиапоптотические гены с ATF4, при этом ингибируя побочные эффекты ATF4, что позволяет предположить профилактический эффект и время вмешательства Nrf2-опосредованной терапии с использованием фитохимических веществ, активирующих Nrf2, такие как сульфорафан, или лекарства, такие как диметилфумарат (рис.). Кроме того, активация ATF4 может приводить к чистому переносу цитозольного НАДФН в митохондрии в постмитотических клетках, таких как сердце и нейроны, что приводит к увеличению НАДФ + / НАДФН в цитозоле (рис.). Сопутствующая активация Nrf2 катализирует реакцию цитозольного NADP + на NADPH, регулируя пентозофосфатный путь и яблочные ферменты. (95) В дополнение к вышеупомянутым механизмам, Nrf2 также регулирует экспрессию гена ATF4 в раковых клетках и косвенно регулирует путь синтеза серина в раковых клетках, что приводит к плохому прогнозу у пациентов с немелкоклеточным раком легкого. (105) Nrf2 может также регулировать MTHFD2, поскольку нокдаун Nrf2 подавляет экспрессию гена MTHFD2 в клетках рака легкого A549. (106) Однако физиологические роли механизмов все еще нуждаются в дальнейшей проверке в будущем.
Заключительные замечания
В этой обзорной статье мы обсудили роль и механизм активации ATF4 во время прогрессирования заболевания и стрессов окружающей среды, а также его роль ниже внешних сигналов роста. Мы предполагаем, что путь ISR-ATF4 представляет собой реакцию выживания против митохондриального стресса, а ATF4 отвечает за гомеостатические и митогормезные реакции у млекопитающих.
Благодарности
Мы благодарим доктора Джунсей Мимура за критическое прочтение и предложения по рукописи. Эта работа была поддержана номером гранта MEXT / JSPS KAKENHI (26111010) и грантом на институциональные исследования Университета Хиросаки.
Аббревиатуры
| AARE | аминокислотно-ответный элемент | |||
| ATF4 | активирующий фактор транскрипции 4 | |||
| ASNS | аспарагин-синтетаза кальций-синтетаза | кальций-синтетаза | ||
| CARE | Ответный элемент C / EBP-ATF | |||
| Cn | кальцинейрин | |||
| CnAβ1 | кальцинейрин каталитическая субъединица β1 | |||
| CO19 10 | ||||
| Фактор сборки CO19 субъединица цитохром с оксидазы 5B | ||||
| 4E-BP | eIF4E-связывающий белок | |||
| eIF2α | фактор эукариотической инициации 2α | |||
| FGF | 907 | 907 FGF-FGF | D-фактор роста | -Friedroblast |
| общий контроль недерепрессируемый-2 | ||||
| Glut4 | переносчик глюкозы 4 | |||
| HRI | гем-регулируемый ингибитор трансляции | |||
| ISR | интегрированный стрессовый ответ LEGO | Наследственная оптическая нейропатия | ||
| MAM | митохондриально-ассоциированная мембрана | |||
| MEF | эмбриональный фибробласт мыши | |||
| MELAS | митохондриальная миопатия 9012 R192 9752-миопатия, миопатия 9192, энцефалопатия, энцефалопатия 947 миоклоническая эпилепсия с рваными красными волокнами | |||
| mETC | митохондриальная электронная транспортная цепь | |||
| MRS | ретроградная передача сигналов митохондрий | |||
| MURE | элемент | | митохондриальный UPR ответ 52 | нейрогенная атаксия и пигментный ретинит | |
| OxPhos | оксидативное фосфорилирование | |||
| PEO | прогрессирующая внешняя офтальмоплегия | |||
| PERK12R-2-рецидивирующая 975-альфа-9-AR-9-AR-9-AR-9-9-R-9-R-9-R-9-R-9-R-9-R-9-R-9-R-9-R-9-C-90-2 -подобный эндулоплазматический | ||||
| PKR | dsRNA-активированная протеинкиназа | |||
| ROS | реактивные формы кислорода | |||
| TFAM | фактор митохондриальной транскрипции A | |||
| TOR197 9075 цель rapam2 гомолог 3 | ||||
| UCP1 | разобщающий белок 1 | |||
| uORF | вышестоящие открытые рамки считывания | |||
| UPR | развернутый ответ белка | 0
l Конфликты интересов были раскрыты.
Ссылки
1. Лейн Н. Митонуклеарное соответствие: улучшение физической формы и плодовитости на протяжении поколений способствует старению внутри поколений. Биологические исследования. 2011; 33: 860–869. [PubMed] [Google Scholar] 2. Уоллес, округ Колумбия. Митохондриальная парадигма метаболических и дегенеративных заболеваний, старения и рака: начало эволюционной медицины. Анну Рев Жене. 2005; 39: 359–407. [Бесплатная статья PMC] [PubMed] [Google Scholar] 3. Морено-Лошуэртос Р., Ацин-Перес Р., Фернандес-Силва П. и др. Различия в продукции активных форм кислорода объясняют фенотипы, связанные с распространенными вариантами митохондриальной ДНК мышей.Нат Жене. 2006. 38: 1261–1268. [PubMed] [Google Scholar] 4. Frechin M, Enkler L, Tetaud E, et al. Экспрессия ядерных и митохондриальных генов, кодирующих АТФ-синтазу, синхронизируется путем разборки мультисинтетазного комплекса. Mol Cell. 2014; 56: 763–776. [PubMed] [Google Scholar] 5. Кувийон М.Т., Сото IC, Шипковенская Г., Чёрчман Л.С. Синхронизированные программы митохондриальной и цитозольной трансляции. Природа. 2016; 533: 499–503. [Бесплатная статья PMC] [PubMed] [Google Scholar] 6. Арно Т., Мишель С., Ренар П.Ретроградная передача сигналов митохондрий и UPR mt: Где мы находимся у млекопитающих? Int J Mol Sci. 2015; 16: 18224–18251. [Бесплатная статья PMC] [PubMed] [Google Scholar] 9. да Кунья FM, Торелли NQ, Ковальтовски AJ. Митохондриальная ретроградная передача сигналов: триггеры, пути и результаты. Oxid Med Cell Longev. 2015; 2015: 482582. [Бесплатная статья PMC] [PubMed] [Google Scholar] 10. Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, et al. Соответствие митохондриальной и ядерной ДНК формирует метаболизм и здоровое старение.Природа. 2016; 535: 561–565. [PubMed] [Google Scholar] 11. Бутов Р.А., Авадхани Н.Г. Передача сигналов митохондрий: ретроградный ответ. Mol Cell. 2004; 14: 1–15. [PubMed] [Google Scholar] 12. Магасаник Б, Кайзер CA. Регулирование содержания азота в Saccharomyces cerevisiae . Ген. 2002; 290: 1–18. [PubMed] [Google Scholar] 14. Ляо X, Бутов Р.А. RTG1 и RTG2: два дрожжевых гена, необходимые для нового пути коммуникации от митохондрий к ядру. Клетка. 1993; 72: 61–71. [PubMed] [Google Scholar] 15. Лю З., Бутов Р.А.Переключение транскрипции в экспрессии генов цикла трикарбоновых кислот дрожжей в ответ на снижение или потерю дыхательной функции. Mol Cell Biol. 1999; 19: 6720–6728. [Бесплатная статья PMC] [PubMed] [Google Scholar] 16. Лю З., Секито Т., Эпштейн CB, Бутов РА. RTG-зависимая передача сигналов от митохондрий к ядру негативно регулируется белком Lst8p с семью WD-повторами. EMBO J. 2001; 20: 7209–7219. [Бесплатная статья PMC] [PubMed] [Google Scholar] 18. Наргунд AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM.Накопление фактора транскрипции ATFS-1 в митохондриях и ядрах способствует восстановлению OXPHOS во время UPR (mt) Mol Cell. 2015; 58: 123–133. [Бесплатная статья PMC] [PubMed] [Google Scholar] 19. Guaragnella N, Butow RA. ATO3, кодирующий предполагаемый наружный переносчик аммония, представляет собой RTG-независимый ретроградный чувствительный ген, регулируемый GCN4 и аминокислотной сенсорной системой Ssy1-Ptr3-Ssy5. J Biol Chem. 2003; 278: 45882–45887. [PubMed] [Google Scholar] 20. Делани Дж. Р., Ахмед У, Чоу А. и др. Стресс-профилирование мутантов долголетия идентифицирует Afg3 как митохондриальную детерминанту трансляции цитоплазматической мРНК и старения.Ячейка старения. 2013; 12: 156–166. [Бесплатная статья PMC] [PubMed] [Google Scholar] 21. Брукс П.С., Юн Ю., Роботэм Дж. Л., Андерс М. В., Шеу СС. Кальций, АТФ и АФК: митохондриальный треугольник любви-ненависти. Am J Physiol Cell Physiol. 2004; 287: C817 – C833. [PubMed] [Google Scholar] 22. Бисвас Г., Адебанджо О.А., Фридман Б.Д. и др. Ретроградная передача сигналов Ca 2+ в скелетных миоцитах C2C12 в ответ на митохондриальный генетический и метаболический стресс: новый способ межорганических перекрестных помех. EMBO J. 1999; 18: 522–533.[Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Гуха М., Тан В., Сондхеймер Н., Авадхани Н. Г.. Роль кальциневрина, hnRNPA2 и Akt в активации транскрипции ядерных генов, опосредованной митохондриальным респираторным стрессом. Biochim Biophys Acta. 2010; 1797: 1055–1065. [Бесплатная статья PMC] [PubMed] [Google Scholar] 24. Рохас Л.М., Сен-Пьер Дж., Ульдри М., Ягер С., Хандшин С., Шпигельман Б.М. Фундаментальная система клеточного энергетического гомеостаза, регулируемая PGC-1alpha. Proc Natl Acad Sci U S. A. 2007; 104: 7933–7938.[Бесплатная статья PMC] [PubMed] [Google Scholar] 25. Хсеу Ю.К., Ло Х.В., Кориви М., Цай Ю.К., Тан MJ, Ян Х.Л. Дерматозащитные свойства эрготионеина за счет индукции Nrf2 / ARE-опосредованных антиоксидантных генов в кератиноцитах человека, облученных УФА. Free Radic Biol Med. 2015; 86: 102–117. [PubMed] [Google Scholar] 26. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Гемоксигеназа-1 регулирует биогенез сердечных митохондрий посредством Nrf2-опосредованного транскрипционного контроля ядерного респираторного фактора-1. Circ Res.2008; 103: 1232–1240. [Бесплатная статья PMC] [PubMed] [Google Scholar] 27. Амери К, Харрис А.Л. Активирующий фактор транскрипции 4. Int J Biochem Cell Biol. 2008; 40: 14–21. [PubMed] [Google Scholar] 28. Бэрд TD, Wek RC. Фосфорилирование эукариотического фактора инициации 2 и трансляционный контроль метаболизма. Adv Nutr. 2012; 3: 307–321. [Бесплатная статья PMC] [PubMed] [Google Scholar] 29. Fujita Y, Ito M, Nozawa Y, Yoneda M, Oshida Y, Tanaka M. Индукция CHOP (гомологичный белок C / EBP) и ASNS (аспарагинсинтетаза) в гибридных клетках, несущих мутации митохондриальной ДНК MELAS и NARP.Митохондрия. 2007; 7: 80–88. [PubMed] [Google Scholar] 30. Cortopassi G, Danielson S, Alemi M и др. Митохондриальное заболевание активирует транскрипты развернутого белкового ответа и клеточного цикла и подавляет везикулярную секрецию и специфичные для олигодендроцитов транскрипты. Митохондрия. 2006; 6: 161–175. [PubMed] [Google Scholar] 31. Суомалайнен А., Маджандер А., Халтиа М. и др. Множественные делеции митохондриальной ДНК в нескольких тканях пациента с тяжелой заторможенной депрессией и семейной прогрессирующей внешней офтальмоплегией.J Clin Invest. 1992; 90: 61–66. [Бесплатная статья PMC] [PubMed] [Google Scholar] 32. Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. Аутосомно-доминантное заболевание с множественными делециями митохондриальной ДНК, начинающимися в области D-петли. Природа. 1989; 339: 309–311. [PubMed] [Google Scholar] 33. Tyynismaa H, Mjosund KP, Wanrooij S, et al. Мутантная митохондриальная геликаза Twinkle вызывает множественные делеции мтДНК и поздние митохондриальные заболевания у мышей. Proc Natl Acad Sci U S. A. 2005; 102: 17687–17692.[Бесплатная статья PMC] [PubMed] [Google Scholar] 34. Tyynismaa H, Кэрролл CJ, Раймундо N и др. Митохондриальная миопатия вызывает реакцию, подобную голоданию. Hum Mol Genet. 2010; 19: 3948–3958. [PubMed] [Google Scholar] 35. Ким К.Х., Чжон Ю.Т., Ким С.Х. и др. Индуцированное метформином ингибирование дыхательной цепи митохондрий увеличивает экспрессию FGF21 за счет активации ATF4. Biochem Biophys Res Commun. 2013; 440: 76–81. [PubMed] [Google Scholar] 36. Кейпер С., Ост М., Чадт А. и др. Вызванное разъединением скелетных мышц долголетие у мышей связано с повышенным метаболизмом субстрата и индукцией системы эндогенной антиоксидантной защиты.Am J Physiol Endocrinol Metab. 2013; 304: E495 – E506. [PubMed] [Google Scholar] 37. Кейперт С., Ост М., Иоганн К. и др. Разобщение митохондрий в скелетных мышцах приводит в движение эндокринные перекрестные помехи за счет индукции FGF21 как миокина. Am J Physiol Endocrinol Metab. 2014; 306: E469 – E482. [PubMed] [Google Scholar] 38. Сильва Дж. М., Вонг А., Карелли В., Кортопасси Г. А. Подавление митохондриальной функции вызывает интегрированный стрессовый ответ в олигодендроглии. Neurobiol Dis. 2009; 34: 357–365. [PubMed] [Google Scholar] 39.Мартинес-Рейес I, Санчес-Араго М., Куэсва Ю.М. AMPK и GCN2-ATF4 сигнализируют о репрессии митохондрий в клетках рака толстой кишки. Биохим Дж. 2012; 444: 249–259. [PubMed] [Google Scholar] 40. Гараева А.А., Ковалева И.Е., Чумаков П.М., Евстафьева А.Г. Дисфункция митохондрий индуцирует экспрессию гена SESN2 посредством активации фактора транскрипции 4. Клеточный цикл. 2016; 15: 64–71. [Бесплатная статья PMC] [PubMed] [Google Scholar] 41. Эндо Дж., Сано М., Катаяма Т. и др. Метаболическое ремоделирование, вызванное митохондриальным альдегидным стрессом, стимулирует толерантность к окислительному стрессу в сердце.Circ Res. 2009; 105: 1118–1127. [PubMed] [Google Scholar] 42. Хуанг М.Л., Сивагурунатан С., Тинг С. и др. Молекулярные и функциональные изменения в модели сердца мышей с атаксией Фридрейха: активация интегрированного стрессового ответа, фосфорилирование eIF2α и индукция нижестоящих мишеней. Am J Pathol. 2013; 183: 745–757. [PubMed] [Google Scholar] 43. Ким К. Х., Чон Й. Т., О Х и др. Дефицит аутофагии приводит к защите от ожирения и инсулинорезистентности за счет индукции Fgf21 как митокина. Nat Med.2013; 19: 83–92. [PubMed] [Google Scholar] 44. Celardo I, Lehmann S, Costa AC, Loh SH, Miguel Martins L. Регулирование dATF4 митохондриального фолат-опосредованного одноуглеродного метаболизма является нейрозащитным. Смерть клетки отличается. 2017; 24: 638–648. [Бесплатная статья PMC] [PubMed] [Google Scholar] 45. Себастьян Д., Эрнандес-Альварес М.И., Сегалес Дж. И др. Митофузин 2 (Mfn2) связывает функцию митохондрий и эндоплазматического ретикулума с передачей сигналов инсулина и необходим для нормального гомеостаза глюкозы. Proc Natl Acad Sci U S A.2012; 109: 5523–5528. [Бесплатная статья PMC] [PubMed] [Google Scholar] 46. Перейра Р.О., Тадинада С.М., Засадный Ф.М. и др. Дефицит OPA1 способствует секреции FGF21 из мышц, что предотвращает ожирение и инсулинорезистентность. EMBO J. 2017; 36: 2126–2145. [Бесплатная статья PMC] [PubMed] [Google Scholar] 47. Houtkooper RH, Mouchiroud L, Ryu D и др. Дисбаланс митонуклеарных белков как консервативный механизм долголетия. Природа. 2013; 497: 451–457. [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Мишель С., Канонн М., Арно Т, Ренар П.Ингибирование экспрессии митохондриального генома запускает активацию CHOP-10 посредством передачи сигналов клетки, зависящей от интегрированного стрессового ответа, но не от ответа митохондриального развернутого белка. Митохондрия. 2015; 21: 58–68. [PubMed] [Google Scholar] 49. Bruning A, Brem GJ, Vogel M, Mylonas I. Тетрациклины вызывают зависимую от клеточного стресса активацию ATF4 и ингибирование mTOR. Exp Cell Res. 2014; 320: 281–289. [PubMed] [Google Scholar] 50. Bao XR, Ong SE, Goldberger O и др. Дисфункция митохондрий изменяет одноуглеродный метаболизм в клетках человека.Элиф. 2016; 5. pii: elo575. [Бесплатная статья PMC] [PubMed] [Google Scholar] 51. Мартинус Р.Д., Гарт Г.П., Вебстер Т.Л. и др. Селективная индукция митохондриальных шаперонов в ответ на потерю митохондриального генома. Eur J Biochem. 1996; 240: 98–103. [PubMed] [Google Scholar] 52. Мунк С., Харпер Дж. У. Ответ митохондриального развернутого белка контролирует процессинг и трансляцию матричной пре-РНК. Природа. 2016; 534: 710–713. [Бесплатная статья PMC] [PubMed] [Google Scholar] 53. Боуман Л., Шлирф А., Лутц А. К. и др. Паркин транскрипционно регулируется ATF4: свидетельство взаимосвязи между митохондриальным стрессом и стрессом ER.Смерть клетки отличается. 2011; 18: 769–782. [Бесплатная статья PMC] [PubMed] [Google Scholar] 54. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Мембраны, ассоциированные с митохондриями: состав, молекулярные механизмы и физиопатологические последствия. Антиоксидный окислительно-восстановительный сигнал. 2015; 22: 995–1019. [PubMed] [Google Scholar] 55. Джо Й., Ким С., Ким Х. Дж. И др. FGF21-индуцированный монооксидом углерода опосредует метаболический гомеостаз через путь PERK / ATF4. FASEB J. 2018; 32: 2630–2643. [Бесплатная статья PMC] [PubMed] [Google Scholar] 56.Бейкер Б.М., Наргунд А.М., Сан Т., Хейнс С.М. Защитное соединение митохондриальной функции и синтеза белка через киназу eIF2α GCN-2. PLoS Genet. 2012; 8: e1002760. [Бесплатная статья PMC] [PubMed] [Google Scholar] 57. Ван С.Ф., Чен М.С., Чжоу Ю.С. и др. Дисфункция митохондрий усиливает устойчивость к цисплатину в клетках рака желудка человека через путь GCN2-eIF2α-ATF4-xCT, активируемый ROS. Oncotarget. 2016; 7: 74132–74151. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Ян Ц., Саддерт Дж., Данг Т., Бачу Р.М., Макдональд Дж. Г., ДеБерардини Р.Дж.Клеткам глиобластомы требуется глутаматдегидрогеназа, чтобы выжить при нарушениях метаболизма глюкозы или передачи сигналов Akt. Cancer Res. 2009; 69: 7986–7993. [Бесплатная статья PMC] [PubMed] [Google Scholar] 60. Салливан Л. Б., Гуй Д. Ю., Хосиос А. М., Буш Л. Н., Фрейнкман Е., Вандер Хайден М. Г.. Поддержка биосинтеза аспартата является важной функцией дыхания пролиферирующих клеток. Клетка. 2015; 162: 552–563. [Бесплатная статья PMC] [PubMed] [Google Scholar] 61. Ким И., Сюй В., Рид Дж. Гибель клеток и стресс эндоплазматического ретикулума: актуальность болезни и терапевтические возможности.Nat Rev Drug Discov. 2008; 7: 1013–1030. [PubMed] [Google Scholar] 62. Лионаки Э., Маркаки М., Паликарас К., Тавернаракис Н. Митохондрии, аутофагия и возрастные нейродегенеративные заболевания: новые взгляды на сложное взаимодействие. Biochim Biophys Acta. 2015; 1847: 1412–1423. [PubMed] [Google Scholar] 64. Дакер Г.С., Чен Л., Моршер Р.Дж. и др. Обращение цитозольного одноуглеродного потока компенсирует потерю митохондриального фолатного пути. Cell Metab. 2016; 23: 1140–1153. [Бесплатная статья PMC] [PubMed] [Google Scholar] 65.Круг А.К., Гутбир С., Чжао Л. и др. Транскрипционная и метаболическая адаптация нейронов человека к митохондриальному токсиканту MPP + . Cell Death Dis. 2014; 5: e1222. [Бесплатная статья PMC] [PubMed] [Google Scholar] 66. Ян Дж. Х., Вада А., Йошида К. и др. Специфическая для мозга делеция Phgdh показывает ключевую роль биосинтеза l-серина в контроле уровня d-серина, коагониста рецептора N -метил-d-аспартата, во взрослом мозге. J Biol Chem. 2010; 285: 41380–41390. [Бесплатная статья PMC] [PubMed] [Google Scholar] 67.Jun DY, Park HS, Lee JY и др. Положительная регуляция промоторной активности гена 3-фосфоглицератдегидрогеназы человека (PHGDH) опосредуется факторами транскрипции Sp1 и NF-Y. Ген. 2008; 414: 106–114. [PubMed] [Google Scholar] 68. Никканен Дж., Форсстрём С., Euro L и др. Дефекты репликации митохондриальной ДНК нарушают клеточные пулы dNTP и модифицируют одноуглеродный метаболизм. Cell Metab. 2016; 23: 635–648. [PubMed] [Google Scholar] 69. Бен-Сахра I, Ходжадж Дж., Рикуль С.Дж., Асара Дж. М., Мэннинг Б.Д. mTORC1 индуцирует синтез пурина посредством контроля цикла митохондриального тетрагидрофолата.Наука. 2016; 351: 728–733. [Бесплатная статья PMC] [PubMed] [Google Scholar] 70. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Количественный анализ потока показывает, что производство НАДФН зависит от фолиевой кислоты. Природа. 2014; 510: 298–302. [Бесплатная статья PMC] [PubMed] [Google Scholar] 71. Пискунова Е., Агатоклеус М., Мерфи М.М. и др. Окислительный стресс подавляет отдаленные метастазы клетками меланомы человека. Природа. 2015; 527: 186–191. [Бесплатная статья PMC] [PubMed] [Google Scholar] 72. Андерсон Д.Д., Кинтеро С.М., Стовер П.Дж.Идентификация пути биосинтеза тимидилата de novo в митохондриях млекопитающих. Proc Natl Acad Sci U S. A. 2011; 108: 15163–15168. [Бесплатная статья PMC] [PubMed] [Google Scholar] 73. Моршер Р.Дж., Дакер Г.С., Ли Ш. и др. Трансляция митохондрий требует фолат-зависимого метилирования тРНК. Природа. 2018; 554: 128–132. [Бесплатная статья PMC] [PubMed] [Google Scholar] 74. Падрон-Барте Л., Вильяльба-Ореро М., Гомес-Салинеро Дж. М. и др. Активация серинового одноуглеродного метаболизма кальциневрином Aβ1 снижает гипертрофию миокарда и улучшает функцию желудочков.J Am Coll Cardiol. 2018; 71: 654–667. [PubMed] [Google Scholar] 75. Ким KH, Ли MS. FGF21 как гормон стресса: роль FGF21 в адаптации к стрессу и лечении метаболических заболеваний. Diabetes Metab J. 2014; 38: 245–251. [Бесплатная статья PMC] [PubMed] [Google Scholar] 76. Lehtonen JM, Forsström S, Bottani E, et al. FGF21 является биомаркером нарушений митохондриальной трансляции и поддержания мтДНК. Неврология. 2016; 87: 2290–2299. [Бесплатная статья PMC] [PubMed] [Google Scholar] 77. Доган С.А., Пуджол С., Маити П. и др.Тканеспецифическая потеря DARS2 активирует стрессовые реакции независимо от дефицита дыхательной цепи в сердце. Cell Metab. 2014; 19: 458–469. [PubMed] [Google Scholar] 78. Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Фактор роста фибробластов 21 регулирует энергетический метаболизм, активируя путь AMPK-SIRT1-PGC-1alpha. Proc Natl Acad Sci U S. A. 2010; 107: 12553–12558. [Бесплатная статья PMC] [PubMed] [Google Scholar] 79. Хочак Э., Сабо В., Кальман Н. и др. Ингибирование PARP защищает митохондрии и снижает продукцию ROS через ретроградный путь PARP-1-ATF4-MKP-1-MAPK.Free Radic Biol Med. 2017; 108: 770–784. [PubMed] [Google Scholar] 81. Леверенц Дж., Бакстер П., Кассубек Р. и др. Фосфоинозитид-3-киназы активируют систему xc (-) через фактор инициации эукариот 2α и активирующий фактор транскрипции 4 — путь, активный при глиобластомах и эпилепсии. Антиоксидный окислительно-восстановительный сигнал. 2014; 20: 2907–2922. [Бесплатная статья PMC] [PubMed] [Google Scholar] 82. Park Y, Reyna-Neyra A, Philippe L, Thoreen CC. mTORC1 уравновешивает предложение клеточных аминокислот с потребностью в синтезе белка посредством посттранскрипционного контроля ATF4.Cell Rep. 2017; 19: 1083–1090. [Бесплатная статья PMC] [PubMed] [Google Scholar] 83. Идзумия Ю., Бина Х.А., Оучи Н., Акасаки Ю., Харитоненков А., Уолш К. FGF21 является Akt-регулируемым миокином. FEBS Lett. 2008. 582: 3805–3810. [Бесплатная статья PMC] [PubMed] [Google Scholar] 84. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR контролирует окислительную функцию митохондрий через транскрипционный комплекс YY1-PGC-1alpha. Природа. 2007; 450: 736–740. [PubMed] [Google Scholar] 85. Биджур Г.Н., Йопе Р.С.Быстрое накопление Akt в митохондриях после активации фосфатидилинозитол-3-киназы. J Neurochem. 2003. 87: 1427–1435. [Бесплатная статья PMC] [PubMed] [Google Scholar] 86. Хан Н.А., Никканен Дж., Яцуга С. и др. mTORC1 регулирует митохондриальную интегрированную стрессовую реакцию и прогрессирование митохондриальной миопатии. Cell Metab. 2017; 26: 419–428.e415. [PubMed] [Google Scholar] 87. Фелкин Л.Е., Нарита Т., Гермак Р. и др. Вариант сплайсинга кальциневрина кальциневрин Aβ1 улучшает сердечную функцию после инфаркта миокарда, не вызывая гипертрофии.Тираж. 2011; 123: 2838–2847. [PubMed] [Google Scholar] 88. Лара-Пецци Э., Винн Н., Пол А. и др. Встречающийся в природе вариант кальциневрина подавляет активность FoxO и усиливает регенерацию скелетных мышц. J Cell Biol. 2007. 179: 1205–1218. [Бесплатная статья PMC] [PubMed] [Google Scholar] 89. Гуха М., Сринивасан С., Бисвас Г., Авадхани Н.Г. Активация нового пути рецептора инсулиноподобного фактора роста-1, опосредованного кальциневрином, измененный метаболизм и инвазия опухолевых клеток в клетки, подвергшиеся митохондриальному респираторному стрессу.J Biol Chem. 2007. 282: 14536–14546. [Бесплатная статья PMC] [PubMed] [Google Scholar] 90. Мурата Х., Сакагути М., Джин И и др. Новый цитозольный путь киназы, связанной с болезнью Паркинсона, BRPK / PINK1: активация AKT через mTORC2. J Biol Chem. 2011; 286: 7182–7189. [Бесплатная статья PMC] [PubMed] [Google Scholar] 91. Каземи С., Мунир З., Балцис Д. и др. Новая функция киназ eIF2alpha как индукторов сигнального пути фосфоинозитид-3 киназы. Mol Biol Cell. 2007. 18: 3635–3644. [Бесплатная статья PMC] [PubMed] [Google Scholar] 92.Раджеш К., Кришнамурти Дж., Казимерчак У. и др. Фосфорилирование фактора инициации трансляции eIF2α по серину 51 определяет клеточные судьбы Akt в ответ на окислительный стресс. Cell Death Dis. 2015; 6: e1591. [Бесплатная статья PMC] [PubMed] [Google Scholar] 93. Хан Дж., Бэк Ш., Хур Дж. И др. Регуляция транскрипции, вызванная ER-стрессом, увеличивает синтез белка, что приводит к гибели клеток. Nat Cell Biol. 2013; 15: 481–490. [Бесплатная статья PMC] [PubMed] [Google Scholar] 94. Евстафьева А.Г., Гараева А.А., Хуторненко А.А. и др.Устойчивая недостаточность митохондриального респираторного комплекса III вызывает апоптотическую гибель клеток за счет p53-опосредованного ингибирования активности активирующего фактора транскрипции, способствующей выживанию. 4. Смерть клеток Dis. 2014; 5: e1511. [Бесплатная статья PMC] [PubMed] [Google Scholar] 95. Ито К., Мимура Дж., Ямамото М. Открытие отрицательного регулятора Nrf2, Keap1: исторический обзор. Антиоксидный окислительно-восстановительный сигнал. 2010; 13: 1665–1678. [PubMed] [Google Scholar] 96. Ито К., Йе П, Мацумия Т., Танджи К., Одзаки Т.Возникающие функциональные перекрестные помехи между системой Keap1-Nrf2 и митохондриями. J Clin Biochem Nutr. 2015; 56: 91–97. [Бесплатная статья PMC] [PubMed] [Google Scholar] 97. Сулиман HB, Carraway MS, Tatro LG, Piantadosi CA. Новая активирующая роль CO в биогенезе сердечных митохондрий. J Cell Sci. 2007; 120: 299–308. [PubMed] [Google Scholar] 98. Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M. NRF2 регулирует экспрессию PINK1 в условиях окислительного стресса. PLoS One. 2015; 10: e0142438. [Бесплатная статья PMC] [PubMed] [Google Scholar] 99.Wu KLH, Wu CW, Chao YM, Hung CY, Chan JYH. Нарушение регуляции Nrf2 митохондриального биогенеза в ростральном вентролатеральном мозговом веществе при гипертензии, вызванной системным воспалением. Free Radic Biol Med. 2016; 97: 58–74. [PubMed] [Google Scholar] 100. Пуллиам Д.А., Дипа С.С., Лю И и др. Мыши Surf1 (- / -), дефицитные по комплексу IV, инициируют митохондриальные стрессовые реакции. Биохим Дж. 2014; 462: 359–371. [Бесплатная статья PMC] [PubMed] [Google Scholar] 101. He CH, Gong P, Hu B и др. Идентификация активирующего фактора транскрипции 4 (ATF4) как белка, взаимодействующего с Nrf2.Значение для регуляции гена гемоксигеназы-1. J Biol Chem. 2001; 276: 20858–20865. [PubMed] [Google Scholar] 102. Е П, Мимура Дж, Окада Т. и др. Nrf2- и ATF4-зависимая активация xCT модулирует чувствительность клеток карциномы мочевого пузыря Т24 к ингибированию протеасом. Mol Cell Biol. 2014; 34: 3421–3434. [Бесплатная статья PMC] [PubMed] [Google Scholar] 103. Zong ZH, Du ZX, Li N и др. Роль Nrf2 и ATF4 в дифференциальной индукции CHOP путем ингибирования протеасом в раковых клетках щитовидной железы.Biochim Biophys Acta. 2012; 1823: 1395–1404. [PubMed] [Google Scholar] 104. Zong ZH, Du ZX, Zhang HY и др. Участие Nrf2 в индукции ORP150, опосредованной ингибированием протеасом, в раковых клетках щитовидной железы. Oncotarget. 2016; 7: 3416–3426. [Бесплатная статья PMC] [PubMed] [Google Scholar] 105. ДеНикола GM, Чен PH, Малларки Э. и др. NRF2 регулирует биосинтез серина при немелкоклеточном раке легкого. Нат Жене. 2015; 47: 1475–1481. [Бесплатная статья PMC] [PubMed] [Google Scholar] 106. Мицуиси Ю., Тагучи К., Каватани Ю. и др.Nrf2 перенаправляет глюкозу и глутамин в анаболические пути при метаболическом перепрограммировании. Раковая клетка. 2012; 22: 66–79. [PubMed] [Google Scholar] 107. Ву З., Пигсервер П., Андерссон Ю. и др. Механизмы, контролирующие митохондриальный биогенез и дыхание через термогенный коактиватор PGC-1. Клетка. 1999. 98: 115–124. [PubMed] [Google Scholar] 108. Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Активация АМФ-активированной протеинкиназы увеличивает количество митохондриальных ферментов в скелетных мышцах.J. Appl Physiol (1985) 2000; 88: 2219–2226. [PubMed] [Google Scholar] 109. Zhang F, Pracheil T, Thornton J, Liu Z. Аденозинтрифосфат (АТФ) является кандидатной сигнальной молекулой в пути ретроградного ответа от митохондрий к ядру. Гены (Базель) 2013; 4: 86–100. [Бесплатная статья PMC] [PubMed] [Google Scholar]Atf-6 регулирует продолжительность жизни посредством ER-митохондриального гомеостаза кальция
https://doi.org/10.1016/j.celrep.2020.108125Получить права и контентОсновные моменты
- •
-
Утрата посредника UPR ER ATF-6 в C.elegans увеличивает продолжительность жизни
- •
-
ATF-6 обращает вспять возрастное увеличение кальретикулина, поглотителя кальция ER
- •
-
Отток кальция ER через InsP 3 R требуется для увеличения продолжительности жизни
- •
-
Высвобождение кальция ER увеличивает митохондриальную динамику и биоэнергетику
Резюме
По отдельности дисфункция как эндоплазматического ретикулума (ER), так и митохондрий связана со старением, но как коммуникация между этими органеллами может быть направлена на способствовать долголетию неясно.Здесь мы представляем доказательства того, что у Caenorhabditis elegans ингибирование медиатора консервативного развернутого белка (UPR ER ), активирующего фактор транскрипции ( atf ) -6 , увеличивает продолжительность жизни за счет модуляции гомеостаза кальция и передачи сигналов на митохондрии. Atf-6 Потеря обеспечивает долголетие за счет подавления кальциевого буфера ER, кальретикулина. Высвобождение кальция ER через рецептор инозитолтрифосфата (IP 3 R / itr-1 ) необходимо для долговечности, в то время как прирост функции IP 3 R / itr-1 достаточен для увеличения продолжительности жизни.Подчеркивая координацию между органеллами, митохондриальный канал импорта кальция mcu-1 также необходим для долголетия atf-6 . Ингибирование IP 3 R приводит к нарушению биоэнергетики митохондрий и гиперфузии, чего достаточно для подавления долгой жизни у мутантов atf-6 . Это исследование показывает важность обращения с кальцием в органеллах как критического результата для UPR ER при определении качества старения.
Ключевые слова
InsP3R
UPR
старение
долголетие
кальретикулин
межорганеллярная коммуникация
Рекомендуемые статьиЦитирующие статьи (0)
© 2020 Автор (ы).
Рекомендуемые статьи
Ссылки на статьи
Существенная роль митохондриальной функции в синтезе адипонектина в адипоцитах | Диабет
Настоящее исследование демонстрирует, что функция митохондрий важна для производства адипонектина в адипоцитах. Лечение розиглитазоном и меры, увеличивающие биогенез митохондрий (Ad-NRF-1), увеличивают синтез адипонектина. Напротив, химические вещества, которые нарушают функцию митохондрий, и меры, снижающие биогенез митохондрий (mtTFA-specific siRNA), снижают синтез адипонектина.Эти эффекты розиглитазона или митохондриальной активности были специфичны для адипонектина, так как лечение на другие адипоцитокины либо не влияло, либо изменялось в противоположном направлении. Наши данные также демонстрируют, что нарушение функции митохондрий вызывает стресс ER, который, в свою очередь, активирует серию реакций по снижению транскрипции адипонектина.
Как обсуждалось ранее, адипоциты подвергаются созреванию в два этапа: дифференцировка и гипертрофия (6).Вполне возможно, что усиление функции митохондрий необходимо для дифференцировки адипоцитов, поскольку для этого требуется много энергии. Вновь дифференцированные клетки с повышенным содержанием митохондрий будут небольшими по размеру, поскольку повышенное окисление жирных кислот снизит накопление внутриклеточных триглицеридов. Хорошо известно, что уровни синтеза адипонектина увеличиваются в маленьких жировых клетках (6), и настоящие данные показывают, что это связано с усилением функции митохондрий. Напротив, в гипертрофированных жировых клетках синтез адипонектина снижается из-за нарушения функции митохондрий.Количество гипертрофированных жировых клеток увеличивается у людей с ожирением, что объясняет снижение уровня адипонектина в плазме. Пока неясно, почему продукция адипонектина специфически регулируется митохондриальной функцией. Однако, поскольку адипоциты производят огромное количество адипонектина по сравнению с другими белками (31), синтез адипонектина может быть важным энергоемким процессом в адипоцитах. Если это так, то вполне логично, что синтез адипонектина жестко регулируется клеточной системой производства энергии (т.е. митохондриальная функция).
Процессы обработки кальция и сворачивания белка в ER требуют большого количества АТФ, а депривация глюкозы и гипоксия — условия, истощающие клеточный АТФ — хорошо известные стимулы, вызывающие стресс ER (23). В этом отношении удивительно, что мало известно о том, как изменения в функции митохондрий влияют на функцию ER. В нашем исследовании нарушение функции митохондрий вызвало стресс ER, и это было обращено вспять за счет увеличения митохондриального биогенеза.Нарушение функции митохондрий последовательно активирует JNK и ATF3, оба из которых, как было показано, активируются стрессом ER (28,32). Ингибирование JNK и ATF3 обращает вызванное митохондриальной дисфункцией снижение транскрипции адипонектина, демонстрируя, что митохондриальная дисфункция снижает транскрипцию адипонектина с помощью ряда молекулярных механизмов, включая стресс ER и активацию JNK и ATF3.
Chevillotte et al.(33) недавно сообщили, что олигомицин и антимицин A снижают экспрессию адипонектина за счет увеличения продукции активных форм кислорода (ROS) и экспрессии CHOP10, что ингибирует экспрессию гена адипонектина, вмешиваясь в область связывания CCAAT / энхансер-связывающий белок промотора гена адипонектина. Поскольку митохондрии являются одной из основных органелл продукции АФК, особенно когда ее функция нарушена (34), существует вероятность того, что дисфункция митохондрий снижает синтез адипонектина за счет увеличения производства АФК (33,35).Значение этого потенциального механизма в связывании функции митохондрий с синтезом адипонектина требует непосредственного рассмотрения в будущих исследованиях.
Предыдущие исследования выявили многочисленные гормоны и факторы транскрипции, участвующие в синтезе адипонектина (16,26,36). Среди них TNF-α и интерлейкин-6, как хорошо известно, снижают синтез адипонектина (36), и считается, что повышенные уровни этих адипоцитокинов способствуют снижению уровня адипонектина при ожирении.В настоящем исследовании мы использовали TNF-α в качестве цитокина, который снижает биогенез митохондрий и увеличивает восприимчивость к митохондриальным повреждениям (18,19), но мы не можем исключить возможность того, что некоторые из его эффектов не зависели от его влияния на митохондрии. Точно так же влияние розиглитазона на синтез адипонектина может включать, по крайней мере частично, механизмы, независимые от митохондрий: было показано, что рецепторы PPARγ / ретиноида X напрямую связываются с функциональным PPAR-чувствительным элементом в промоторе адипонектина человека и увеличивают промотор активность (26).
Функция митохондрий в скелетных мышцах считается важным фактором, определяющим чувствительность к инсулину всего тела (37,38). Недавнее исследование показало, что адипонектин увеличивает количество митохондрий и их функцию в скелетных мышцах за счет активации 5-AMP-активированной протеинкиназы (39), которая, как известно, стимулирует биогенез митохондрий скелетных мышц в ответ на хроническую депривацию энергии или тренировки на выносливость (40). Если это так, то очень интересно обнаружить, что синтез адипонектина регулируется функцией митохондрий в адипоцитах.Таким образом, существует вероятность того, что митохондриальная (или метаболическая) активность в адипоцитах регулирует митохондриальную (или метаболическую) активность и действие инсулина в скелетных мышцах через адипонектин. Эта концепция согласуется с предыдущими исследованиями, демонстрирующими, что метаболические изменения в жировой ткани влияют на действие инсулина в скелетных мышцах (41).
В заключение, функция митохондрий связана с синтезом адипонектина в адипоцитах, а нарушение функции митохондрий в жировой ткани может объяснить снижение уровня адипонектина в плазме при ожирении.Нарушение функции митохондрий активирует ряд механизмов, включая стресс ER, JNK и ATF3, для снижения синтеза адипонектина. Нарушение функции митохондрий (37,38,42,43) и стресс ER (27–29,44) были предложены отдельно как важные детерминанты инсулинорезистентности в различных тканях субъектов с ожирением. Ввиду текущего открытия последовательных изменений митохондриальной функции и стресса ER в жировой ткани, необходимы дальнейшие исследования, чтобы изучить, происходят ли эти отдельные механизмы также последовательно в других тканях, участвующих в патогенезе инсулинорезистентности.
Перед печатью опубликовано на сайте http://diabetes.diabetesjournals.org 7 сентября 2007 г. DOI: 10.2337 / db07-0510.
E.H.K., J.-Y.P. и H.-S.P. внес равный вклад в эту работу.
Дополнительную информацию к этой статье можно найти в онлайн-приложении по адресу http://dx.doi.org/10.2337/db07-0510.
Расходы на публикацию этой статьи были частично покрыты за счет оплаты страницы. Поэтому данная статья должна быть помечена как «реклама» в соответствии с 18 U.S.C. Раздел 1734 исключительно для указания этого факта.
Экспериментальное метилирование ДНК, нацеленное на митохондрии, идентифицирует метилирование GpC, а не метилирование CpG, как потенциальный регулятор экспрессии митохондриальных генов
Культура клеток
C33A (рак шейки матки человека), HCT116 (рак толстой кишки человека) и HEK293T (эмбриональная почка человека) получено из АТСС. OSE-C2 (иммортализованные эпителиальные клетки яичников человека 43 ), CiGenCs (условно иммортализованные эндотелиальные клетки клубочков человека 44 ), IHH (иммортализованные гепатоциты человека 45 ) и BEAS-2B ρ 0 эпителиальных клеток человека (эпителиальные клетки бронхов человека мтДНК) были любезно предоставлены доктором.Ричард Эдмондсон, доктор Саймон Сэтчелл, доктор Хан Мошаге и доктор Роланд Хоффманн, соответственно.
CiGenCs и IHH культивировали на покрытых желатином колбах, тогда как клетки BEAS-2B ρ 0 культивировали на покрытых коллагеном колбах. Все клеточные линии, за исключением клеток IHH и BEAS-2B ρ 0 , культивировали в среде DMEM с высоким содержанием глюкозы (25 мМ глюкозы) (Lonza) с добавлением 10% FCS (Perbio Hyclone), 2 мМ L-глутамина (BioWhittaker) и 50 мкг / мл сульфата гентамицина (Invitrogen). Кроме того, среда IHH содержала 20 мЕд / мл инсулина (Novo Nordisk) и 50 нмоль / л дексаметазона (Sigma).Клетки BEAS-2B ρ 0 культивировали в среде DMEM с высоким содержанием глюкозы с добавлением 25% FCS, 2 мМ L-глутамина, 1% P / S, 2,5 мкг / мл амфотерицина B (Sigma), 1 × раствор аминокислот MEM. (Sigma), 1 × раствор незаменимых аминокислот MEM (Sigma), витамины (Sigma), 50 мкг / мл уридина (Sigma). Все клетки хранили в увлажненном инкубаторе с 5% CO 2 при 37 ° C.
Для обработки с высоким и низким уровнем глюкозы клетки дважды промывали PBS и культивировали в течение 4 дней либо на низкой (5 мМ), либо на высокой (25 мМ) глюкозе DMEM.
Клонирование
Белки, нацеленные на митохондрии, были клонированы с использованием одной «мастер-синтетической конструкции». Эта «основная синтетическая конструкция» была синтезирована в Bio Basic Canada и содержит (от 5′- до 3′-конца): 1. Последовательность Козака; 2. N-концевой 49-аминокислотный сигнал митохондриальной локализации (MLS) субъединицы F1β митохондриальной АТФ-синтазы 46 ; 3. открыть позицию 1; 4. HA-тег; 5. Гибкий линкер 17-а.о. — (SGGGG) 3 SS 46 ; 6. открытая позиция 2 для эпигенетического фермента; 7.C-концевой 18-аминокислотный сигнал экспорта в ядро (NES) неструктурного белка 2 минутного вируса мышей 47 ; 8. стопкодон. Добавление рестрикционных ферментов между отдельными компонентами обеспечило гибкость в клонировании белков, нацеленных на митохондрии: BamHI — Kozak — MLS — NruI … AvrII — Открытое положение 1 — BsIWI … NruI — HAtag — гибкий линкер — EcoRV … AscI — Открытое положение 2 — PacI … EcoRV — NES — stopcodon — NotI .Эту главную конструкцию субклонировали в pCDH-CMV-MCS-EF1-copGFP (CD511B-1) с использованием сайтов рестрикции BamHI и NotI (System Biosciences). В этой плазмиде EF1-copGFP заменяли устойчивостью к SV40-пуромицину с использованием рестрикционных ферментов NotI и XhoI. Дополнительный NES был клонирован в окончательную конструкцию. Более того, «открытая позиция 1» была удалена с помощью разложения NruI. Для визуализации плазмиды те же рестрикционные ферменты были использованы для субклонирования mCherry в «открытое положение 1». Все конструкции, описанные ниже, были клонированы в эту плазмиду.В качестве отрицательного контроля с использованием расщепления EcoRV была создана конструкция без эффекторного домена (NoED), не содержащая белка в «открытом положении 2».
DNMT1
Для получения продукта ПЦР варианта транскрипта DNMT1, нацеленного на митохондрии (mtDNMT1), из эталонной кДНК человека (Clontech, с произвольным праймированием) или пула кДНК с произвольным праймированием из линий клеток человека (HEK293T, HCT116, HeLa, Использовали IHH, SiHa, Caski, SKOV3, HepG2, C33A, OSE-C2), праймеры ( BclI -mtDNMT1- NotI ), как описано в таблице 1.Амплификация mtDNMT1 оказалась безуспешной, несмотря на использование широкого спектра стратегий: разные ДНК-полимеразы использовались в соответствии с протоколом производителя (высокоточная ДНК-полимераза Phusion (Thermo Scientific), ДНК-полимераза Pfu (Thermo Scientific), ДНК-полимераза Taq (Thermo Scientific)) состав смеси для ПЦР варьировался (тип буфера, концентрация MgCl 2 , добавление ДМСО), различные протоколы ПЦР (температуры плавления, время элонгации, количество циклов и т. Д.) были протестированы. Следовательно, в качестве альтернативы, кодирующая последовательность нормального (не нацеленного на митохондрии) гена DNMT1 (клон кДНК MGC: 161505 IMAGE: 8991943) была получена с использованием праймеров ( AscI -DNMT1- PacI ), как описано в таблице 1. Для достижения митохондрий-нацеливания DNMT1 этот продукт ПЦР был клонирован в pCDH-CMV-master синтетическая конструкция-SV40-puro с использованием сайтов рестрикции AscI и PacI, в результате чего был получен гибкий линкер MLS1x-HAtag-DNMT1-2xNES. .
М.SssI, M.CviPI, hM.CviPII и
E . coli промотор conIIПлазмида, содержащая M.SssI 48 и его каталитически неактивный двойной мутант (E186A, R230A), M.SssI∆∆ 30 , были ранее получены от доктора Antal Kiss. Плазмиды, содержащие M.CviPI 31 и hM.CviPII 32 , были любезно предоставлены доктором Майклом Кладде. До того, как нечеловеческие ДНК-метилтрансферазы были клонированы в синтетическую конструкцию pCDH-CMV-master-SV40-puro, E .Промотор coli conII был включен в обратной ориентации сразу за NES. Это было сделано путем отжига комплементарной пары олигонуклеотидов, содержащих conII и расщепленные фрагменты NotI (таблица 1). Короче говоря, эквимолярные концентрации прямого и обратного олигонуклеотидов смешивали с NEB Buffer 4 и инкубировали на водяной бане при 95 ° C в течение 5 минут. При выключении водяной бани олигонуклеотидам давали медленно остыть до комнатной температуры. Отожженные олигонуклеотиды использовали в последующих процедурах клонирования.Конвергентная транскрипция conII относительно гена ДНК-метилтрансферазы снижает токсичность из-за неплотной экспрессии даже в E . coli штаммов без рестрикции, зависящей от метилирования 49 . Субклонирование M.SssI с использованием рестрикционных ферментов AscI и PacI или продукта ПЦР M.CviPI и hM.CviPII, содержащего сайты рестрикции AscI и PacI, позволило получить синтетическую конструкцию pCDH-CMV-master-conII-SV40-puro, содержащую MLS1x. -HAtag-гибкий линкер- (M.SssI / M.CviPI / hM.CviPII) -2x NES. Все конструкции были подтверждены ПЦР колоний и секвенированием (Baseclear). Трансформацию плазмид, содержащих нечеловеческие DNMT, проводили в E . coli, клеток ER1821, все остальные были выполнены в E . coli Top10 клеток.
Вирусная доставка нацеленных на митохондрии конструкций
Лентивирусные частицы, содержащие нацеленные на митохондрии конструкции, получали, как описано ранее 50 .Вкратце, упаковывающие клетки HEK293T котрансфицировали кальций-фосфатным методом плазмидами, содержащими ATF, и вирусными упаковывающими плазмидами, содержащими gag / pol и белок G вируса везикулярного стоматита в соотношении 3: 2: 1. Вирусный супернатант собирали через 48 и 72 часа после трансфекции и использовали в комбинации с 6 мкг / мл полибрена (Sigma-Aldrich) для заражения клеток-хозяев C33A и HCT116. Через три дня после трансдукции были получены стабильные клеточные линии с использованием отбора 1 мкг / мл пуромицина (Sigma) в течение 5 дней.Селекционную среду обновляли каждые 2 дня.
Праймеры для валидации
Все праймеры, используемые для амплификации мтДНК, были подтверждены на агарозном геле для специфической амплификации мтДНК, а не так называемых NUMT, ядерных копий мтДНК. Для этого в качестве отрицательного контроля использовали ДНК клеток BEAS-2B ρ 0 , не содержащую мтДНК. Для каждой пары праймеров q (RT) -PCR была построена стандартная кривая для расчета эффективности пары праймеров (дополнительный рис. 4).
Количественная ПЦР в реальном времени (qRT-PCR)
Общую РНК выделяли с использованием набора для очистки РНК GeneJET (Thermo Scientific) в соответствии с протоколом производителя, включая дополнительную 15-минутную обработку DNaseI (Roche) для удаления загрязнения ДНК.РНК количественно определяли с использованием спектрофотометра Nanodrop 1000 (Thermo Scientific). 1 мкг РНК подвергали обратной транскрипции в кДНК с использованием случайных гексамерных праймеров с помощью набора для обратной транскрипции QuantiTect (Qiagen) в соответствии с протоколом производителя. Каждая реакция qRT-PCR содержала 500 нМ каждой пары праймеров, 10 нг кДНК и 1xABsolute qPCR SYBR Green, Rox Mix (Thermo Scientific). Праймеры были заново сконструированы, извлечены из банка данных праймеров для ПЦР в реальном времени (RTPrimerDB, http://medgen.urgent.be/rtprimerdb/) или получены из литературы 23, 51, 52 (таблица 1).Реакции qRT-PCR проводили на ViiA7 Real time PCR (Applied Biosystems) в течение 15 минут при 95 ° C, затем 40 циклов по 15 секунд при 95 ° C, 30 секунд при 60 ° C и 30 секунд при 72 ° C. β-актин использовался в качестве гена домашнего хозяйства. Данные и кривые плавления анализировали с использованием программного обеспечения ViiA7 RUO, а относительную экспрессию по сравнению с контролями рассчитывали с использованием метода ∆∆Ct 53 .
Выделение ДНК
Лизис клеток выполняли O / N при 55 ° C в буфере для лизиса TNE (10 мМ Трис / HCl, pH 7.5; 150 мМ NaCl; 10 мМ ЭДТА; 1% SDS) и 100 мкг протеиназы K. На следующий день ДНК выделяли, как описано ранее 50 . Короче говоря, лизированные клетки перемешивали в течение 15 секунд. с насыщенным (6 М) NaCl в соотношении 5: 1. Эту смесь объединяли с равным объемом хлороформ / изоамиловый спирт (24: 1) и перемешивали в течение 60 минут. на роторе с последующим центрифугированием в течение 20 мин при 10000 об / мин при 4 ° C. Тотальную клеточную ДНК (геномную и митохондриальную ДНК) экстрагировали с использованием хлороформа / изоамилового спирта (24: 1), РНКазы A (Thermo Scientific), обработанной в течение 1 ч при 37 ° C, и осаждали с использованием изопропанола.Количество ДНК определяли с помощью спектрофотометра Nanodrop 1000 (Thermo Scientific).
Число копий митохондриальной ДНК (мтДНК) и образование праймера ДНК 7S
10 нг общей клеточной ДНК использовали в качестве исходных данных для количественной ПЦР. Использовали праймеры, амплифицирующие область яДНК (β-актин) и область мтДНК (D-петля) (таблица 1). Для проверки использовали независимую пару праймеров мтДНК области mtCOX1. Реакции количественной ПЦР проводили на ViiA7 Real time PCR (Applied Biosystems) в течение 15 минут при 95 ° C, затем 40 циклов по 15 секунд при 95 ° C, 30 секунд при 60 ° C и 30 секунд при 72 ° C.{\ ast {\ rm {primer}} {\ rm {fficiency}}}) \).
Бисульфитное секвенирование
400 нг ДНК превращали в бисульфит с использованием набора EZ DNA Metylation Gold (Zymo Research) в соответствии с инструкциями производителя. Бисульфитная ПЦР D-петли 13 и mtCOX2 7 была проведена с использованием бисульфит-специфичных праймеров (таблица 1), как описано ранее 7, 13 . Продукты ПЦР клонировали в вектор pCR4-TOPO (Thermo Scientific), и отдельные клоны отправляли на секвенирование.Результаты секвенирования бисульфита анализировали с помощью онлайн-инструмента QUMA (www.quma.cdb.riken.jp/) 55 .
Иммунопреципитация метилированной ДНК (MeDIP)
Для каждой иммунопреципитации 1 мкг общей клеточной ДНК обрабатывали ультразвуком с использованием Bioruptor Pico (20 циклов по 20 дюймов, 40 дюймов). Иммунопреципитацию 5 мкС ДНК проводили с использованием набора для захвата метилированной ДНК Methylamp (Epigentek) в соответствии с инструкциями производителя. Иммунопреципитация ДНК с использованием нормального мышиного антитела IgG выполнялась в качестве отрицательного контроля.Обогащение 5 мС в специфических областях мтДНК анализировали с использованием праймеров для D-петли , mtCYTB , mtCOX2 (как описано ранее в ссылке 18, таблица 1).
Конфокальная микроскопия
Локализацию слитой конструкции M.SssI, нацеленной на mCherry и митохондрии, визуализировали с помощью конфокальной флуоресцентной микроскопии (Leica SP8, линза HC PL APO CS2 63 × / 1,4). Следуя рекомендациям производителя, для окрашивания митохондрий клетки обрабатывали 100 нМ Mitotracker Deep Red FM (Molecular Probes) в течение 30 минут при 37 ° C.Слитый белок M.SssI, нацеленный на mCherry-митохондрии, возбуждали лазерным светом 552 нм, а Mitotracker Deep Red возбуждали лазерным светом 633 нм.
Вестерн-блоттинг
Клетки собирали в буфере для ресуспендирования (100 мМ NaCl, 15 мМ MgCl 2 , 100 мМ Трис, pH 7,5) и инкубировали на льду в течение 10 минут при регулярном встряхивании. Образцы гомогенизировали, промывая клетки 5 раз через иглу G25. Затем фракции ядерных (NER) и митохондриальных (MER) белков собирали с помощью дифференциального центрифугирования 56 .Количественное определение белка выполняли с помощью теста DC BioRad Protein Assay (BioRad). 50 мкг белка загружали в 12% гель SDS-PAGE для обнаружения конструкции, направленной на митохондрии (содержащей HAtag). Блоты блокировали на 1 час 5% обезжиренным молоком в TBS. Для обнаружения первичные антитела инкубировали O / N при 4 ° C, тогда как вторичные антитела инкубировали в течение 1 ч при комнатной температуре. Были использованы следующие антитела: 1: 1000 мышиных анти-HAtag (HA.11, Biolegend), 1: 1000 кроличьих анти-VDAC1 / порин (Ab34726, Abcam), 1: 1000 мышиных антител против ламина B1 (клон L5, Invitrogen). и 1: 1000 конъюгированных с пероксидазой хрена кроличьих антимышиных (P0260, Dako) и свиных антимышиных антител (P0217, Dako).Сигнал вестерн-блоттинга генерировали с помощью субстрата для вестерн-блоттинга Pierce ECL Plus (Thermo Scientific) и детектировали с помощью системы визуализации Biorad ChemiDoc MP (Biorad).
Митохондриальная метаболическая активность
Клетки высевали в 96-луночные планшеты с плотностью 3200 клеток на лунку. Анализ MTS был использован для определения митохондриальной метаболической активности и пролиферации клеток 57 . Короче говоря, через один день (для метаболической активности митохондрий) или через четыре дня (для пролиферации клеток) после посева в каждую лунку добавляли раствор CellTiter 96 A Water One (Promega) и инкубировали в течение 3 часов при 37 ° C.Затем оптическую плотность определяли при 490 нм с помощью считывающего устройства для микропланшетов Versamax (Molecular Devices). При измерении оптической плотности через один день после посева равного количества клеток любое различие в оптической плотности может быть объяснено только разницей в метаболической активности митохондрий, тогда как через четыре дня после посева любая разница в оптической плотности может быть результатом различий в митохондриальной метаболической активности и клеточной активности. пролиферация / жизнеспособность клеток.
Производство митохондриальных АФК
Уровни митохондриального супероксида определяли с помощью зонда MitoSOX Red ROS.Клетки дважды промывали DMEM без фенолового красного и инкубировали с 5 мкМ MitoSOX Red в DMEM без фенолового красного в течение 30 мин при 37 ° C. После обработки клетки трипсинизировали и собирали для измерений FACS (BD LSR-II, BD Biosciences) с использованием УФ-лазера 355 нм в сочетании с фильтром 575/26 нм 58 . В качестве положительного контроля клетки обрабатывали 100 мкМ менадиона в течение 1 ч при 37 ° C.
Анализ гибели клеток
Как описано ранее 59 , чувствительность к гибели клеток, индуцированной ROS, определяли с использованием йодида пропидия (PI) в качестве маркера поздних апоптотических / некротических клеток.Клетки окрашивали в течение 10 мин 5 мкг / мл PI (Sigma-Aldrich) в PBS при 4 ° C в темноте. Флуоресценцию PI измеряли с использованием канала FL-3 проточного цитометра FACScalibur (Beckton Dickenson Biosciences). Процент PI-положительных клеток определяли с помощью программного обеспечения Kaluza 1.2 (Beckman Coulter) и строили графики с использованием программного обеспечения Graphpad Prism 5 (GraphPad Software Inc.).
Статистический анализ
Все эксперименты проводили трижды, если не указано иное. Статистический анализ проводился с использованием программного обеспечения Graphpad Prism 5.Сравнение отдельных групп и множественных групп проводилось с помощью t-критерия Стьюдента или однофакторного дисперсионного анализа с последующим апостериорным критерием Даннета, соответственно. Значение p 0,05 или меньше считалось статистически значимым (* p ≤ 0,05, ** p <0,01 и *** p <0,001).
Многокомпонентный анализ определяет ATF4 как ключевой регулятор митохондриальной стрессовой реакции у млекопитающих | Журнал клеточной биологии
клеток HeLa, обработанных всеми описанными соединениями, дважды промывали ледяным PBS и соскребали в PBS.Осадки клеток лизировали мочевинным буфером (8 М мочевина, 75 мМ NaCl, 50 мМ EPPS, pH 8,5, с добавлением комплексных ингибиторов протеазы [Roche] и ингибиторов фосфатазы PhosSTOP [Roche]). Клеточные остатки центрифугировали в течение 15 минут при 13000 г при 4 ° C. Концентрацию белка определяли с помощью анализа BCA (Thermo Fisher Scientific). 100 мкг лизата восстанавливали 5 мМ TCEP в течение 30 минут, а затем алкилировали 14 мМ йодацетамида в течение 30 минут в темноте, и все реакции инкубировали при комнатной температуре.Затем белки осаждали осаждением метанолом / хлороформом и ресуспендировали в 200 мМ EPPS, pH 8,5. Расщепление белков проводили в течение 16 ч при комнатной температуре с LysC в соотношении 1: 100 (LysC / белок). На следующий день добавляли трипсин в соотношении 1:75 (трипсин / белок) и инкубировали при 37 ° C в течение 5 часов. Затем изобарическое мечение пептидов проводили с использованием реагентов 10-plex TMT (Thermo Fisher Scientific). Вкратце, 100 мкг пептидов метили 0,2 мг реагента TMT (предварительно растворенного в ацетонитриле [ACN]), конечная концентрация ACN ∼30% (об. / Об.) В течение 1 ч при комнатной температуре.Наконец, реакцию гасили на 15 мин гидроксиламином до конечной концентрации 0,3% (об. / Об.). Пептиды, меченные TMT, одинаково объединяли и сушили с помощью вакуумного центрифугирования. Высушенные пептиды ресуспендировали в 3% FA, 3% ACN, обессоливали с использованием картриджа для твердофазной экстракции Sep-Pak C18 (Waters) и снова сушили с помощью центрифугирования в вакууме. Объединенные пептиды, меченные TMT, затем подвергали фракционированию с помощью ВЭЖХ с обращенной фазой с основным pH, как описано (Paulo et al., 2015), и фракционировали на 24 фракции.Половину из них центрифугировали в вакууме досуха, растворяли в 3% FA, 3% ACN, обессоливали с помощью StageTip, снова сушили с помощью вакуумного центрифугирования и ресуспендировали в 10 мкл 3% FA, 3% ACN для масс-спектрометрического анализа. Все образцы обрабатывали на масс-спектрометре Orbitrap Fusion (Thermo Fisher Scientific), соединенном с насосом Proxeon EASY-nLC II LC (Thermo Fisher Scientific). Пептиды фракционировали на микрокапиллярной колонке, заполненной смолой GP-18 (1,8 мкм, 200 Å; Sepax). Пептиды разделяли с использованием 135-минутного градиента 6–26% ACN в 0.125% муравьиная кислота при скорости потока ~ 350 нл / мин. В каждом анализе использовался многоточечный метод TMT на основе MS3 (McAlister et al., 2014). Для каждого спектра FTMS1, обнаруженного в Orbitrap (разрешение 120k; диапазон масс 400–1400 м / z; цель с автоматическим регулированием усиления [AGC] 5 × 10 5 , максимальное время впрыска 100 мс), прекурсоры для анализа MS2 / MS3 были выбраны с использованием параметра TopSpeed, равного 2 с. Анализ MS2 состоял из диссоциации, вызванной столкновениями (квадрупольный анализ ионной ловушки; AGC 8 × 10 3 ; нормализованная энергия столкновения = 35; максимальное время инжекции 150 мс).Чтобы создать ионы-репортеры TMT, сканирование MS3 с синхронным отбором предшественников проводилось на 10 самых интенсивных ионах в спектре MS2. Прекурсоры MS3 с синхронным отбором предшественников были фрагментированы с помощью высокоэнергетической диссоциации, вызванной столкновениями, и проанализированы в Orbitrap (нормализованная энергия столкновения = 55%, AGC = 10 5 , максимальное время впрыска = 150 мс, разрешение = 60 K) . Анализ данных проводился с помощью внутреннего программного обеспечения на основе SEQUEST (Huttlin et al., 2010). Короче говоря, масс-спектры искали в базе данных Uniprot человека (февраль 2014 г.) и в базе данных обратной ловушки.Карбамидометилирование цистеина (+57,0215 D) и метка TMT (+229,1629 D) на остатках лизина и N-концах пептида были добавлены в качестве статических модификаций, а окисление метионина (+15,9949 D) было установлено как вариабельная модификация. Допуск по ионам-предшественникам был установлен на уровне 20 ppm, а толерантность к ионам продукта — на 0,9 D. Соответствие пептидным спектрам было скорректировано до 1% FDR с использованием линейного дискриминантного анализа (Huttlin et al., 2010), количественно оцененного путем извлечения отношения сигнал-шум. соотношение, и белки далее разрушались до конечного уровня FDR на уровне 1% (McAlister et al., 2014; Пауло и др., 2015). Дифференциально экспрессируемые белки были идентифицированы путем подгонки данных к линейной модели и выполнения эмпирического критерия t-статистики с модерацией Байеса, сравнивая каждое стрессовое состояние с контрольными клетками с использованием пакета limma R / Bioconductor (Ritchie et al., 2015). Данные масс-спектрометрической протеомики были депонированы в Консорциум ProteomeXchange (http://proteomecentral.proteomexchange.org) через репозиторий партнеров PRIDE с идентификатором набора данных PXD006293.
Опосредованная mTORC1 активация ATF4 способствует синтезу белка и глутатиона ниже сигналов роста
Существенных изменений:
1) Это исследование идентифицирует перекрытие между ATF4-зависимой экспрессией гена в ответ на пути ISR и mTORC1.Однако анализ не дает понимания лежащего в основе механизма. Что объясняет различные подмножества экспрессии ATF4-зависимых генов в этих условиях? В разделе «Обсуждение» рукописи предполагается, что это может быть связано с различиями в экспрессии ATF4 (высокая или низкая) или с дифференциальным участием гетеродимерных партнеров ATF4, но никаких экспериментальных тестов этих предсказаний не представлено. Разница также может быть связана с комбинаторным действием ATF4 с другими факторами транскрипции на промоторы генов, которые по-разному активируются посредством ISR по сравнению с другими факторами транскрипции.mTORC1. Был бы полезен сравнительный биоинформатический анализ промоторов, соответствующих дифференциально экспрессируемым генам. Более того, в этих условиях требуется биохимический анализ ATF4, включая анализ гетеромерных партнеров и анализ связывания ДНК.
Этот важный вопрос неизвестен для большинства исследований ATF4, который может быть активирован множеством различных вышестоящих стрессовых сигналов, в дополнение к передаче сигналов фактора роста через mTORC1, чтобы вызвать как специфические, так и перекрывающиеся генные ответы.Учитывая, что ATF4-зависимые цели, которые, как было обнаружено, являются общими для передачи сигналов ISR и mTORC1 в нашем анализе, представляют собой многие из канонически сообщаемых целей ATF4 в других условиях, мы предположили и обсудили в нашем первоначальном сообщении, что они могут представлять те цели, которые являются наиболее чувствительными. к индукции уровня белка ATF4. В соответствии с этой идеей, анализ 774 генов, значительно индуцируемых туникамицином ATF4-зависимым образом (рис. 1 — исходные данные 1), показывает, что 61 ген, также значительно индуцируемый инсулином и чувствительный к рапамицину, значительно обогащен среди самых популярных генов. 100 наиболее значимо индуцированы туникамицином (показаны красным на новом рисунке 1E, с отдельными генами, обозначенными на рисунке 1 — исходные данные 1).
New Figure 2: Как было предложено, мы также использовали два биоинформатических подхода для анализа промоторов этих ATF4-зависимых генов-мишеней. Во-первых, мы использовали инструмент CiiiDER для прогнозирования ДНК-связывающих мотивов факторов транскрипции в промоторах 61 ATF4-зависимого гена, общего для передачи сигналов ISR и mTORC1, по сравнению с топ-200 генами ISR only. Это сработало довольно хорошо и показало, что C / EBP-связывающие мотивы являются наиболее обогащенными среди общих генов, тогда как связывающие мотивы для семейства факторов транскрипции TEAD были обогащены только для генов ISR (новый рисунок 2A).Это последнее наблюдение интересно в свете недавних исследований, показывающих, что TEAD и его партнеры по связыванию YAP / TAZ, нижестоящие мишени пути Hippo, активируются стрессом ER и могут взаимодействовать с мишенями UPR (PMID: 25695629; PMID: 31558567). Наш второй беспристрастный анализ использовал базу данных Cistrome опубликованных и курируемых исследований ChIP-seq по всему геному, чтобы определить, были ли обнаружены специфические факторы транскрипции для взаимодействия с промоторами 61 гена с общей регуляцией. Еще раз, среди исследований ChIP-seq было обнаружено, что семейство факторов транскрипции C / EBP наиболее часто связывается с промоторами этих генов (новый рисунок 2B).
ATF4 может гетеродимеризоваться со всем семейством C / EBP (PMID: 12805554). Таким образом, чтобы продолжить наши биоинформатические наблюдения с помощью функциональных анализов, мы использовали siRNA-опосредованный нокдаун отдельных изоформ C / EBP (α, β, δ, γ) и ATF4 и исследовали экспрессию репрезентативных генов из различных функциональных категорий ( Mthfd2 ). , Slc7a5 , Aars ) из списка 61 ATF4-зависимой цели, совместно используемой mTORC1 и ISR в их регулировании. Этот анализ выявил высокую степень перекрестной регуляции между этими различными изоформами и ATF4 в регуляции их собственных транскриптов, что затруднило оценку прямого воздействия одного или нескольких из этих членов семейства C / EBP на нижестоящие мишени (новый рисунок 2C). .К сожалению, мы не смогли идентифицировать надежные изоформ-специфичные антитела C / EBP, которые давали четкие и воспроизводимые сигналы с помощью иммуноблоттинга или IP из лизатов MEF, с антителами, протестированными с использованием siRNA-опосредованного нокдауна в качестве контроля для соответствующих полос. В разной степени C / EBPβ, δ и γ нокдауны снижают экспрессию всех тестируемых мишеней, но только C / EBPγ делает это для каждого гена, не влияя на уровни белка ATF4 (новый рисунок 2C, D). Интересно, что многие из 61 общего идентифицированного гена перекрываются с генами, которые, как было установлено, регулируются ранее с помощью ATF4 посредством гетеродимеризации с C / EBPγ в ответ на стресс, связанный с лишением аминокислот (PMID: 26667036).Таким образом, мы использовали siRNA-опосредованный нокдаун ATF4 или C / EBPγ, чтобы определить влияние этих репрезентативных мишеней на инсулин и рапамицин (новый рисунок 2D). Эти данные продемонстрировали, что C / EBPγ требуется для индукции этих генов инсулином, а также для стимулированного инсулином увеличения экспрессии гена ATF4. Вместе наш новый биоинформатический анализ и функциональные данные предполагают, что участие C / EBPγ отвечает по крайней мере за часть программы экспрессии ATF4-зависимого гена, разделяемой между передачей сигналов mTORC1 и ISR.Однако, как показано при использовании кДНК ATF4, устойчивой к рапамицину, mTORC1-опосредованная регуляция этих генов-мишеней в основном осуществляется за счет воздействия на уровни белка ATF4.
2) Рис. 2A и E. Авторам необходимо объяснить несоответствие между уровнями РНК и белка. Может оказаться полезным денситометрический анализ белков-блотов. Более того, авторы должны показать уровни белка из подгруппы генов «переносчиков аминокислот», как они это делали с генами «зарядки тРНК» и генами «синтеза аминокислот» на рис. 2D и E.
Уровни транскрипта и белка часто не совпадают, учитывая, что на содержание белка независимо влияют различные скорости трансляции, стабильности и деградации. Количественная оценка этих блотов была облегчена использованием системы визуализации LICOR Odyssey Imaging System, которая обеспечивает линейную количественную оценку, а не результаты, полученные с помощью методов блоттинга с ферментной связью. Соответствующее количественное определение белка блотов на рисунке 2D-E теперь представлено в виде нового рисунка 3 — приложения к рисунку 1C-D.Мы протестировали антитела вместе с контролями siRNA на специфичность для переносчиков нескольких аминокислот, которые, как было обнаружено, регулируются на уровне транскрипта с помощью передачи сигналов mTORC1 через ATF4, включая SLC7A5, SLC3A2 / CD98, SLC1A5 и SLC7A11. Некоторые из этих антител работают для блоттинга экстрактов белков человека, но, к сожалению, только SLC7A11 давал специфическую полосу, которая уменьшалась с помощью siRNAs в MEF (новый рисунок 6 — приложение к рисунку 1A), см. Комментарий 5 ниже.
3) MYC контролирует несколько переносчиков аминокислот (PMID: 32022686).Так как MYC может быть активирован ниже mTORC1, авт. Должны исследовать вклад MYC в экспрессию переносчика аминокислот в клетках Tsc2 — / — .
Мы расширили наш анализ генных мишеней ATF4, включив в него переносчики аминокислот ( Slc7a11 , Slc7a5 , Slc3a2 и Slc1a5 ) и гены синтеза незаменимых аминокислот ( Psat1 f и 2 ), некоторые из которых, как было установлено, регулируются Myc в других настройках.Хотя было обнаружено, что некоторые из них значительно уменьшаются с помощью siRNA-опосредованного нокдауна c-Myc, во всех случаях нокдаун ATF4 сильнее и значительно снижает их экспрессию (новый рисунок 3 — рисунок в приложении 1L).
4) Рисунок 3D. Авторам необходимо показать уровни белка мишеней ATF4 (как они это сделали на рисунке 3C), чтобы продемонстрировать устойчивость к рапамицину на уровне белка. Это важно из-за несоответствий между экспрессией РНК и белка, показанной на рис. 2A и E.
Как упоминалось в комментарии 2 выше, мы не ожидаем совпадения экспрессии РНК и белка по степени изменений. Однако с помощью иммуноблоттинга мы подтвердили, что уровни белков, кодируемых конкретными мишенями гена ATF4, действительно повышены и что они устойчивы к рапамицину в нокаутных клетках ATF4, восстановленных с помощью кДНК ATF4, лишенной ее 5’UTR, но не мутантной версии этой кДНК по ATF4-DBD. (новый рисунок 4 — приложение к рисунку 1).
5) Рисунок 5B, C. Авторы должны показать уровни белка Slc7a11 и Slc3a2, чтобы продемонстрировать дифференциальную чувствительность к рапамицину в различных клеточных линиях.
После тестирования нескольких различных коммерчески доступных антител мы смогли найти антитела SLC7a11, которые специфически и надежно распознавали этот белок в экстрактах белков клеток мыши или человека, а также антитело SLC3A2 / CD98, которое работает с белками человека. Подтверждение SiRNA антитела SLC7A11 в MEF представлено на новом рисунке 6 — добавление к рисунку 1A. В соответствии с регулируемыми mTORC1 изменениями уровней транскриптов, описанными в линиях мышей и людей в исходной рукописи, мы обнаружили, что ингибиторы mTOR значительно снижают уровни белка SLC7A11 в Tsc2 — / — MEF (новый рисунок 6D и количественно на новом рисунке. 6 — рисунок в приложении 1B), а в MEF дикого типа инсулин стимулирует увеличение белка SLC7A11 способом, который чувствителен к ингибиторам mTOR (новый рисунок 6E и количественно определен на новом рисунке 6 — рисунок в приложении 1C).Уровни белка SLC7A11, но не SLC3A2 / CD98, также значительно чувствительны к ингибиторам mTOR в человеческих клетках LNCaP и PC3 (новый рисунок 6F и рисунок 6 — приложение к рисунку 1F-H). Как указано выше, эти блоты были количественно определены с использованием системы LICOR.
6) Рисунок 5E. Авторы заключают, что существует критическая роль mTORC1 в контроле поглощения цистина через ATF4. Однако добавление цистина полностью восстанавливает рост клеток, истощенных по ATF4. Как это возможно, если уровни транспортера будут снижены из-за отсутствия ATF4?
Существует очевидная путаница между цистеином (Cys) и цистином (Cys 2 ), отраженная в этом комментарии.Фигура (теперь фигура 6H) и сопровождающий текст описывают восстановление пролиферации цистеином, но не цистином, что также наблюдается на фигуре 7F для уровней глутатиона. Как отмечено в тексте, цистеин попадает в клетки через переносчики нейтральных аминокислот, которые не регулируются ATF4, как это происходит с SLC7A11-зависимым транспортом цистина.
7) На рис. 6Н авторы утверждают, что уровни глутатиона восстанавливались в клетках Atf4 — / — при экзогенной экспрессии ATF4 посредством активации Slc7a11.Однако ATF4 также регулирует субъединицы уровней глутамат-цистеинлигазы (PMID: 17297441). Каковы уровни Gclc и Gclm в ячейках Atf4 — / — на рисунках 6G и H? Авторы должны измерить эти белки в общем ATF4-KO вместо использования siRNA (Рисунок 7 — приложение к рисунку 1E).
В соответствии с нашими данными, показанными в отношении чувствительной к рапамицину экспрессии транскриптов Gclc и Gclm, на которые не влиял siRNA-опосредованный нокдаун ATF4 (Рисунок 7 — рисунок в приложении 1D-E), только незначительные изменения в уровнях белка GCLC и GCLM были обнаружены с помощью Нокаут ATF4 в клетках дикого типа или нокаут и восстановление в Tsc2 — / — клетках (Фиг.7 — приложение к рисунку 1F-G).Инсулин стимулировал умеренное повышение уровней этих белков как в MEF дикого типа, так и в нокаутных по ATF4, способом, чувствительным к ингибиторам mTOR, но опосредованные mTORC1 эффекты на эти белки не были очевидны в клетках Tsc2 — / — с ATF4 или без него.
