
| 1. |
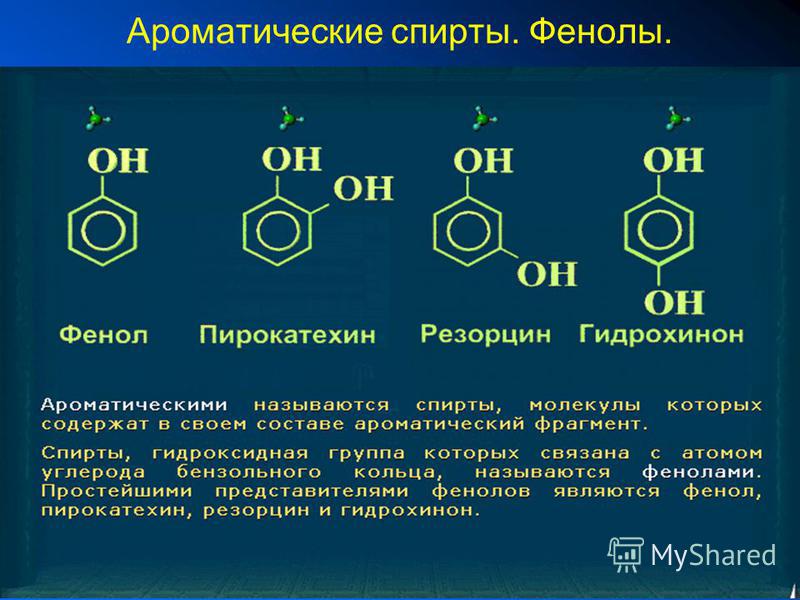
Ароматические спирты и фенолы
Сложность: лёгкое |
1 |
| 2. |
Физические свойства фенола
Сложность: лёгкое |
1 |
3.
|
С чем реагирует фенол
Сложность: лёгкое |
1 |
| 4. |
Фенол и метанол
Сложность: среднее |
2 |
5.
|
Реакции фенолов
Сложность: среднее |
2 |
| 6. |
Продукты реакций
Сложность: среднее |
2 |
7.
|
Кислотные свойства соединений
Сложность: сложное |
3 |
| 8. |
Цепочка превращений
Сложность: сложное |
3 |
9.
|
Задача на смесь
Сложность: сложное |
4 |
обобщение « Спирты и фенолы»
Урок — обобщение « Спирты и фенолы»
(для группы с углубленным изучением химии)
Тип урока: обобщение и закрепление знаний по классу спиртов и фенолов.
Цели урока:
1.
Систематизировать
знания студентов о составе, строении и свойствах органических соединений,
содержащих гидроксильную (спиртовую) функциональную группу, продолжить развитие
представления о генетической связи органических соединений и о химических
реакций.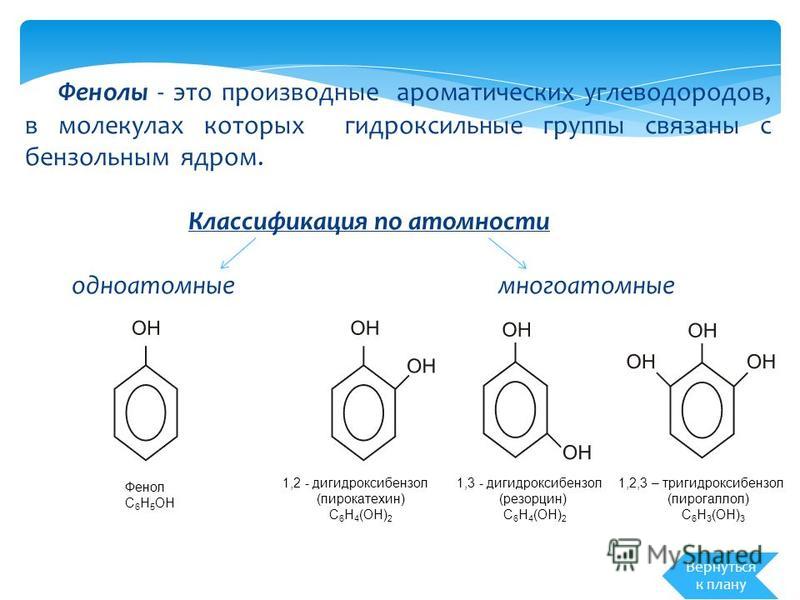
2. Совершенствовать технику безопасности при работе с химическими реактивами, умение соблюдать ее.
3. Способствовать позитивной работы в командах и индивидуально, продолжить развивать коммуникативную способность студента в группе
4. Индивидуальная работа каждого студента на мыслительную , логическиую, рассудительную, анализируемую деятельности собственной и командной на уроке, умение подытоживать свой результат и работы команды.
Методы: поисковый, наглядный, практический, словесный, контрольный.
Реактивы: водные растворы этилового спирта, раствор глицерина, водный раствор фенола, хлорида железа трехвалентного, гидроксида меди двухвалентного, медная проволока
Оборудование: спиртовка, держатель, штатив с пробирками, халат, перчатки.
Дидактический
материал: оценочный
лист каждого ребенка для взаимоконтроля, карточки-кружки желтого, красного,
зеленого цвета, дидактитические карточки, цветные карандаши.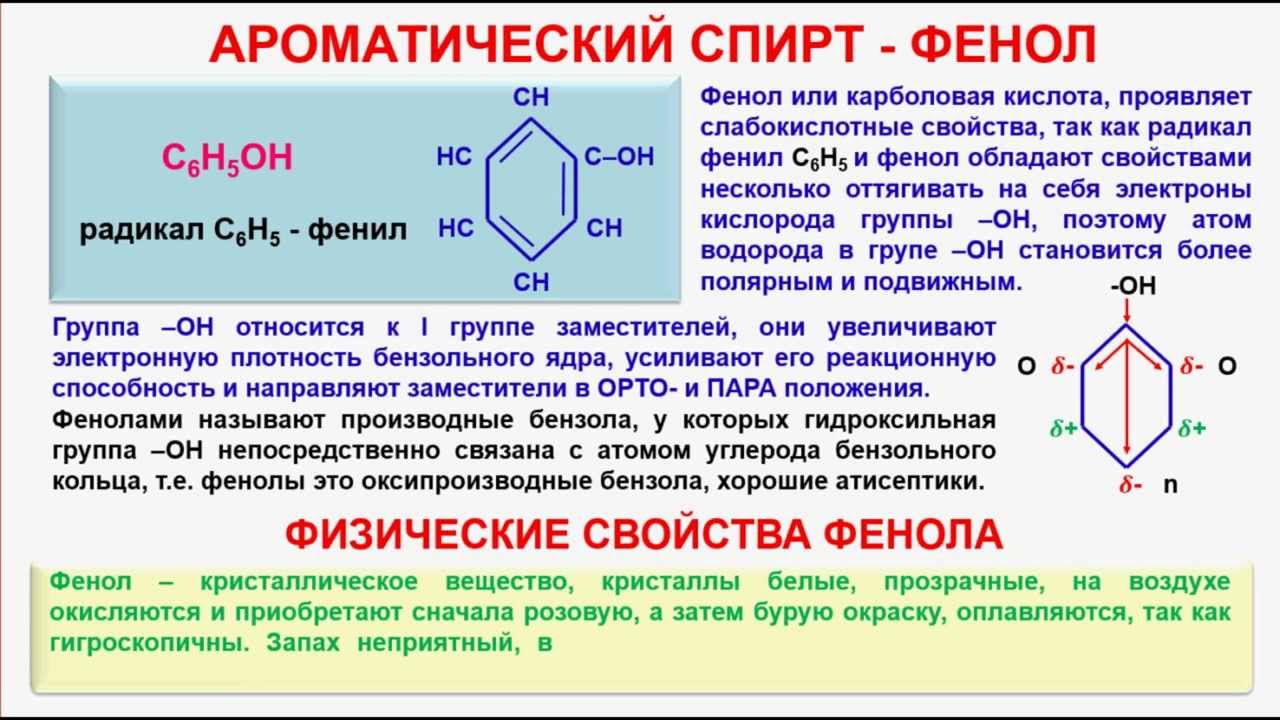
Методическое обеспечение: проектор, интерактивная доска, компьютер, презентация к уроку
Ход урока:
1. Организационный момент. Приветствие учеников, отметка отсутствующих.
2. Постановка цели и задач урока. Мотивация учебной деятельности учащихся.
Учитель: Сегодня мы с вами проведем урок – обобщение на тему «Спирты и фенолы» (учащиеся записывают тему урока), познакомимся с неизвестными, интересными фактами о спиртах и фенолах, вспомним важные моменты в этих темах, проведем опыты и будем работать в командах и индивидуально. Предлагаю назвать свои команды, которые будут соревноваться в интеллектуальных знаниях (вписывают свою фамилию и команду на оценочном листе). За каждый правильный ответ команды ставим + напротив задания, за нераскрытый ответ +-, за неправильный ответ -. Персональный ответ засчитываем в пользу команды.
3.Актуализация знаний.
3. 1.
Ассоциации
по названию команды (командное задание).
1.
Ассоциации
по названию команды (командное задание).
3.2. Практическое задание (командное задание).
3.2.1. Органолептические показатели
Учитель: Перед вами имеются 3 пронумерованных раствора, используя знания о физических свойствах спиртов и фенола, попробуйте по органолептическим показателям определить какие перед вами вещества.
|
Номер пробирки/ вещество |
Органолептические показатели (примерные ответы студентов) |
|
1 пробирка/ (этиловый спирт) |
Жидкость, без цвета, с характерным алкогольным запахом |
|
2 пробирка/ (глицерин) |
Вязкий, густой, тягучий, с желтоватым оттенком, без запаха |
|
3 пробирка/ (фенол) |
Водный раствор с запахом гуаши |
3. 2.2.Качественные
реагенты
2.2.Качественные
реагенты
Учитель: Мы с вами определили по физическим показателям, что за вещества перед вами. Теперь вспомним, какими реагентами вы можете это подтвердить. (Примерные ответы учеников).
Этиловый спирт – оксидом меди двухвалентным (черный налет на медной проволоке очищается до красной цвета меди, появляение легкого запаха яблока, характерный для уксусного альдегида)
Глицерин – гидроксидом меди двухвалентным (ярко фиолетовый цвет глицерата меди двухвалентоного)
Фенол — хлоридом меди трехвалентным (темно фиолетовый цвет)
3.3. Химический диктант «Проверь команду»
1. Формула метилового спирта?
2. К какому классу спиртов относится этиленгликоль по числу функциональной группы?
3. Общая формула предельных одноатомных спиртов?
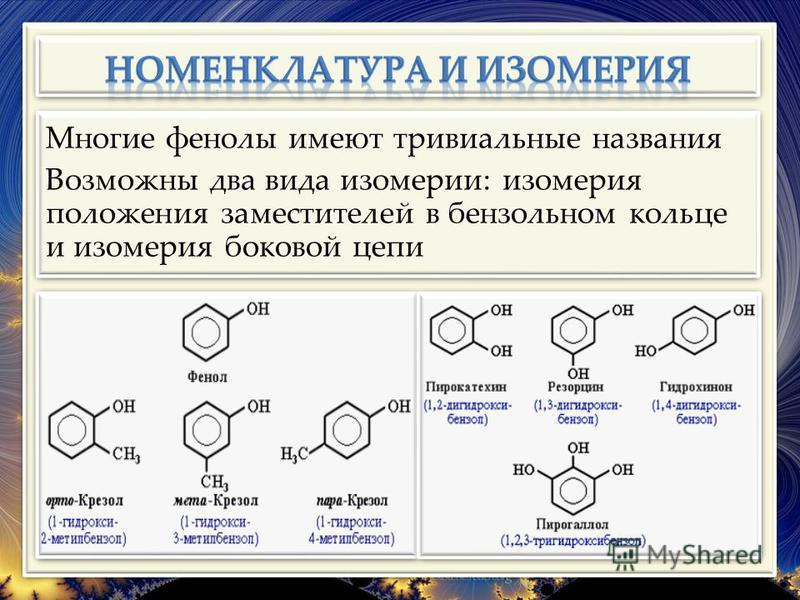
4. Как называется класс кислородсодержащих углеводородов, где гидроксильная группа непосредственно связана с ароматическим радикалом?
5. Валентность углерода в органических соединениях?
6. Формулы
реактивов для обнаружения фенола?
Формулы
реактивов для обнаружения фенола?
7. Алкоголяты – это …
8. Его называют древесным спиртом?
9. Как называется ароматический радикал в феноле?
10. Что такое изомеры? Какая изомерия характерна для одноатомных спиртов?
3.4. Суждения
Учитель: Проанализировать в командах данное суждение и представить ответ, вторая команда слушает ответ, если считают правильным поднимают красный кружок, сомневаются-желтый, если считают неправильный – зеленый, комментируют ответ при необходимости.
1. Все спирты горят. Метан горит, следовательно, метан — это спирт.
2. Вещества обладающие одинаковым качественным и количественным составом, но разным строением называются изомерами. Пропанол – 2 и метилэтиловый эфир обдадают общей формулой: С3Н8О, но разным строением, следовательно, пропанол и диметиловый эфир – изомеры.
3.
Одноатомные
спирты хорошо растворимы в воде, глицерин – спирт, следовательно, глицерин
растворяют в воде в неограниченных количествах.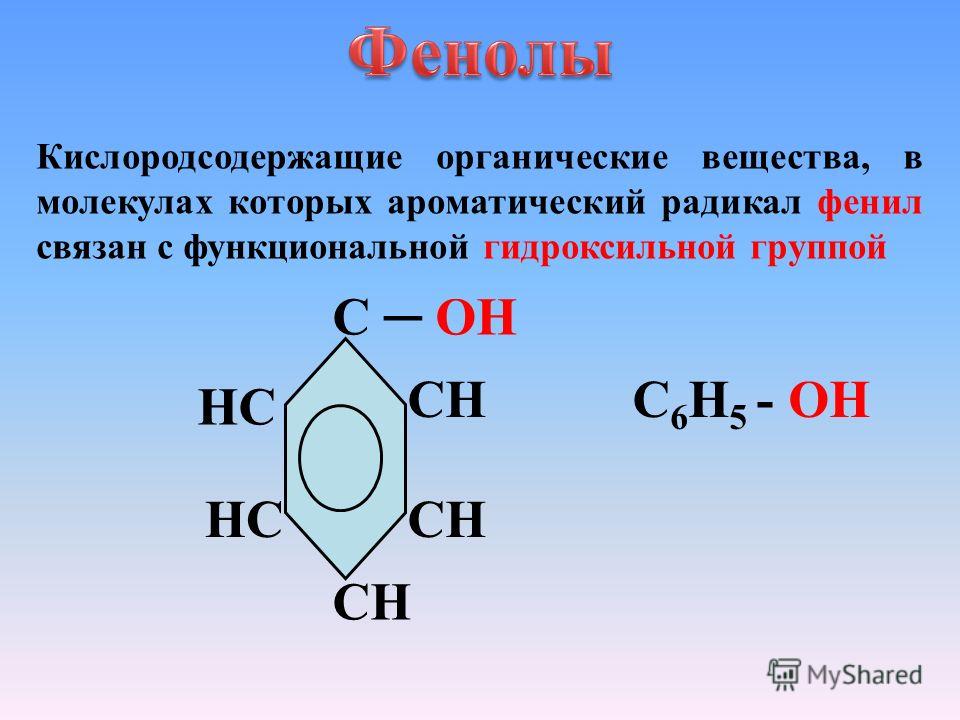
4. Гидроксильная группа связана в органических соединениях водородной связью, следовательно, в феноле присутствует водородная связь.
3.5. Физкультминутка
Очень химию мы любим!
Шеей влево, вправо крутим.
Воздух – это атмосфера,
Если правда, топай смело.
В атмосфере есть азот,
Делай вправо поворот.
Так же есть и кислород,
Делай влево поворот,
Благородные есть газы.
Мы попрыгаем по классу.
Чем выше вверх, тем воздух реже.
Друг другу улыбнулись нежно!
3.6. интересные факты о спиртах и фенолах. Студенты заранее имеют подготовленный доклад. Все слушают внимательно. В конце говорят о практическом применении.
1 доклад.
В природе спирты встречаются в свободным виде.
Вещества также являются компонентами сложных эфиров. Естественный процесс
брожения содержащих углеводы продуктов создает этанол, а также бутанол-1,
изопропанол. Спирты в хлебопекарной промышленности, пивоварении, виноделии
связано с использованием процесса брожения в этих отраслях. Большая часть
феромонов насекомых представлена
спиртами.
Большая часть
феромонов насекомых представлена
спиртами.
Спиртовые производные углеводов в природе:
· сорбит — содержится в ягодах рябины, вишни, имеет сладкий вкус.
Многие растительные душистые вещества — это терпеновые спирты:
· фенхол — компонент плодов фенхеля, смол хвойных деревьев
· борнеол — составной элемент древесины борнеокамфорного дерева
· ментол — компонент состава герани и мяты
Желчь человека, животных содержит желчные многоатомные спирты:
· миксинол
· химерол
· буфол
· холестанпентол
2 доклад:
Глицерин — пищевая добавка Е422 — обеспечивает соединение несмешиваемых жидкостей. Его используют при изготовлении кондитерских, макаронных, хлебобулочных изделий. Глицерин входит в состав ликеров, придает напиткам вязкость, сладкий вкус.
Применение глицерина благоприятно влияет на продукцию:
· клейкость макарон уменьшается
· консистенция конфет, кремов улучшается
· предотвращается быстрое зачерствение хлеба, проседание шоколада
· выпекание изделий происходит без налипания крахмала
Распространено использование спиртов как
сахарозаменителей.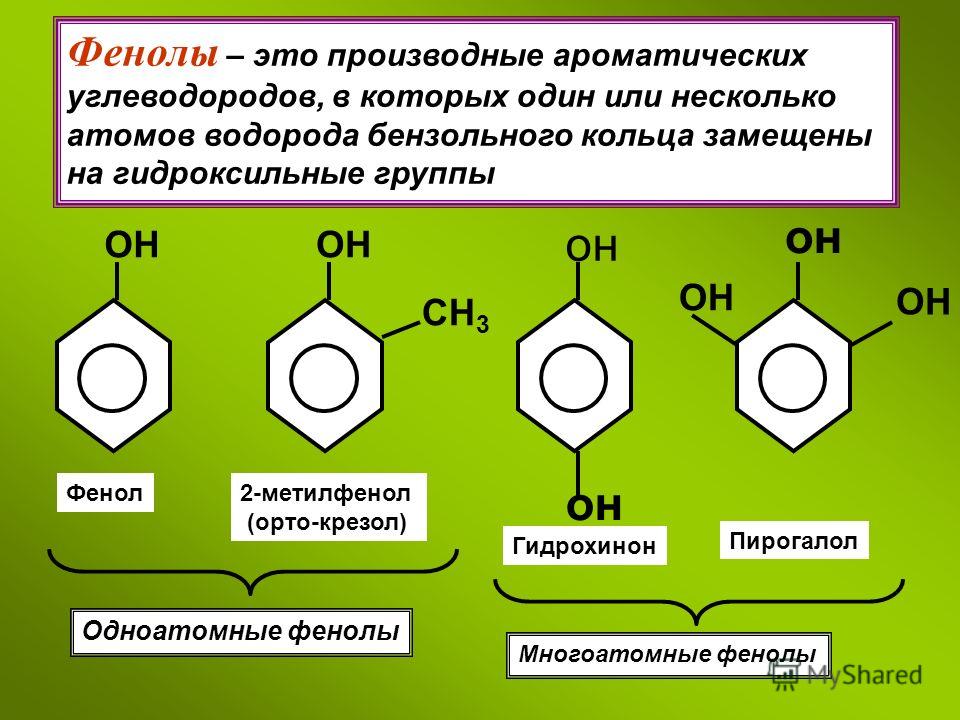 Для этого по свойствам подходят маннит, ксилит, сорбит.
Для этого по свойствам подходят маннит, ксилит, сорбит.
3 доклад.
Открытие фенола Фридлиб Фердинанд Рунге (1794 – 1867 гг) обнаружил в продуктах перегонки каменноугольной смолы белое кристаллическое вещество с характерным запахом гуаши. Ему не удалось определить состав вещества. Огюст Лоран (1807 – 1853 гг) определил состав вещества. Новое вещество обладало выраженными кислотными свойствами и было производным открытого незадолго до этого бензола. Лоран называл бензол «феном», поэтому новая кислота получила название фениловой. Шарль Фредерик Жерар (1816 – 1856 гг) считал полученное вещество спиртом и предложил называть его фенолом. Было установлено, что целая группа веществ обладает подобным строением и свойствами, поэтому их назвали «фенолами».
3.6. Лишнее понятие
Учитель:
Из
перечисленных слов определите лишнее понятие. Вторая команда слушает ответ,
если считают правильным — поднимают красный кружок, сомневаются — желтый, если
считают неправильный – зеленый, комментируют ответ, комментируют ответ при
необходимости.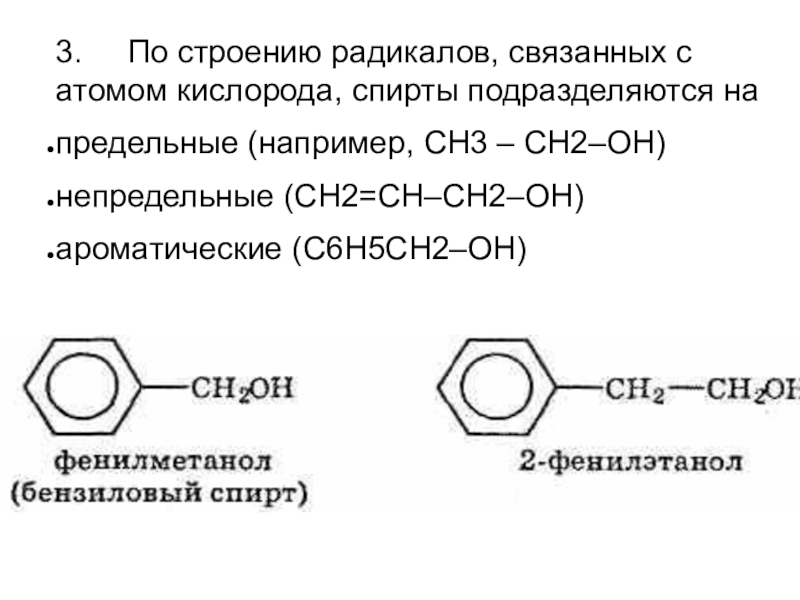
1. Этанол, водородная связь, пропен, бутанол – 1, фенол;
2. Дегидратация, дегидрирование, окисление, этиленгликоль, этерификация;
3. Систематическая, заместительная, межклассовая, тривиальная;
4. Бензол, фенол, метанол, бутанол.
3.7. Синтез — цепочка
Учитель: на выданных дидактических карточках представлены цепочки превращений, используя известные вам уравнения, осуществите синтезы в нескольких стадиях. На команду одну цепочку.
1. Метаналя из карбида алюминия
2. Ацетальдегида из оксида углерода (II).
Учитель: После выполнения задания, студенты сверяются и комментируют ответы.
4. Подведение итогов. Рефлексия.
Цель: формирование у учащихся способности подводить итоги урока, обобщать, делать выводы, характеризовать свои действия
Учитель: лист с отметками
для взаимоконтроля отдаем соседу. Теперь подсчитаем ваши плюсы и минусы, если
плюсов больше чем минусов ставим 5 баллов за работу на уроке, если одинаково –
4 балла.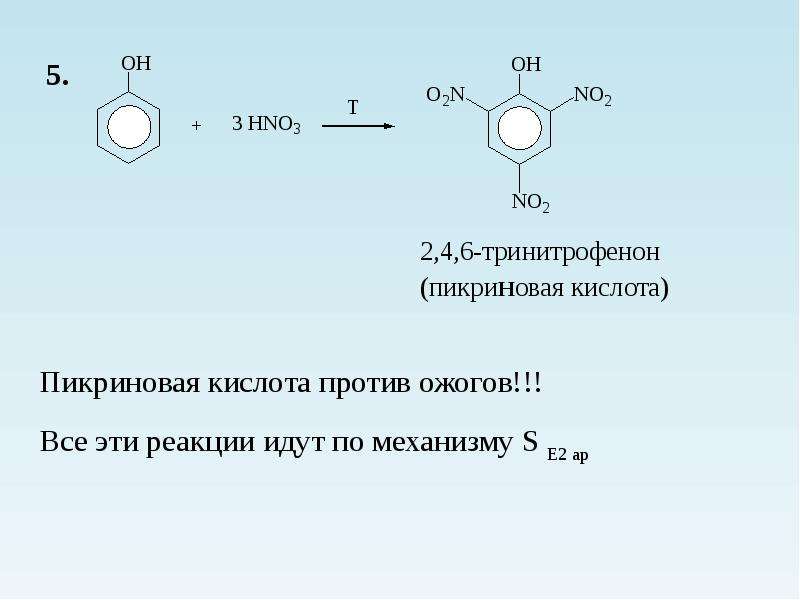
Учитель: вам предлагается оценить урок химии на цветных кружочках, зарисуйте свое настроение.
5. Всем спасибо за урок!
Урок по теме «Фенол»
Государственное автономное профессиональное
образовательное учреждение Пензенской области
«Пензенский многопрофильный колледж»
Отделение строительства.
Методическая разработка конспекта урока
по теме: «Фенол, его строение, свойства и применение».
Выполнила преподаватель: Пивкина Н.В.
г. Пенза.
Конспект урока по теме: «Фенол, его строение, свойства и применение»
1. Образовательные задачи: изучить состав, строение, свойства фенола, рассмотреть зависимость взаимного влияния атомов в молекуле фенола на его свойства, познакомить учащихся с физическими и химическими свойствами фенола, изучить качественную реакцию на фенол, рассмотреть применение фенола и его соединений, их биологическую роль
2. Развивающие задачи: продолжать развивать умение наблюдать, анализировать, делать выводы при выполнении химического эксперимента
Развивающие задачи: продолжать развивать умение наблюдать, анализировать, делать выводы при выполнении химического эксперимента
3. Воспитательные задачи: продолжить формирование химической картины мира через химическую картину природы (познаваемость), расширить представление учащихся о влиянии фенолсодержащих промышленных отходов и строительных материалов на окружающую среду и здоровье человека, рассмотреть биологическую роль фенола и его соединений на организм человека (положительную и отрицательную)
Тип урока: урок — изучения новых знаний.
Методы обучения: словесный, наглядный, практический (химический эксперимент).
Средства обучения: компьютер, проектор, школьный химический эксперимент презентация, видеоролики.
Оборудование и реактивы: демонстрационный эксперимент: растворы С6Н5ОН, NaOH, FeCl3, бромная вода, Na, пробирки, резиновые пробки.
План урока.
1. Организационный момент
2. Актуализация знаний
3. Изучение новых знаний
4. Закрепление
5. Домашнее задание.
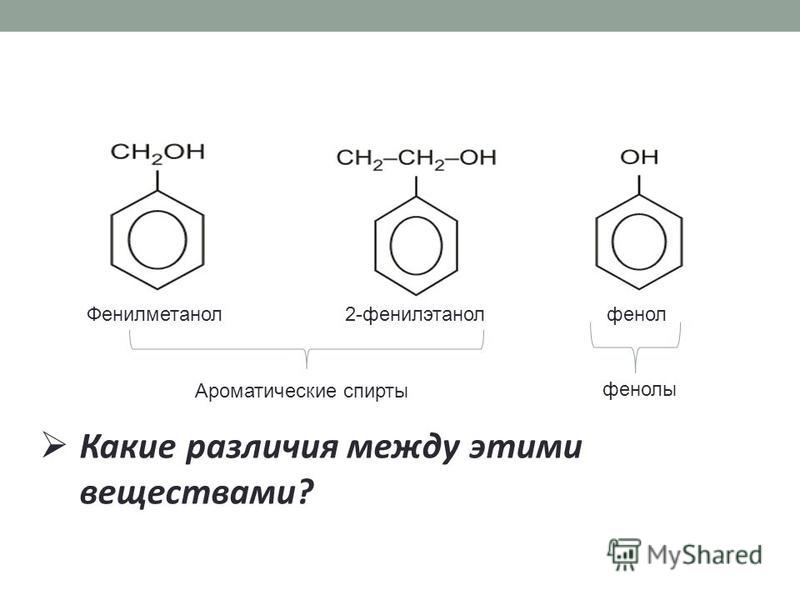
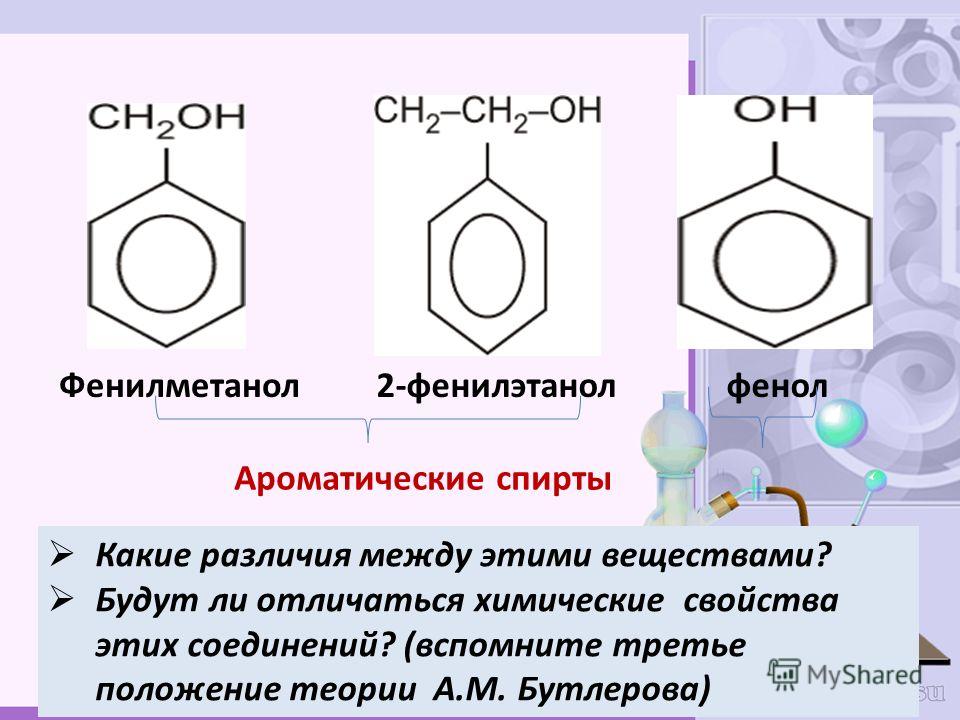
2. Мы с Вами изучили одноатомные и многоатомные спирты.
Вопрос. Какая функциональная группа входит в состав данных соединений?
Сегодня мы познакомимся с веществами, которые тоже содержат данную функциональную группу. Это фенолы. Запишите тему урока.
Цели урока: мы изучим на примере первого представителя данного класса состав и строение фенолов, их свойства и познакомимся с применением соединений данного класса.
3. Формула первого представителя данного класса: С6Н5ОН.
Называется вещество фенол. Другое его название – карболовая кислота.
Посмотрите на структурные формулы: фенола и других представителей данного класса.
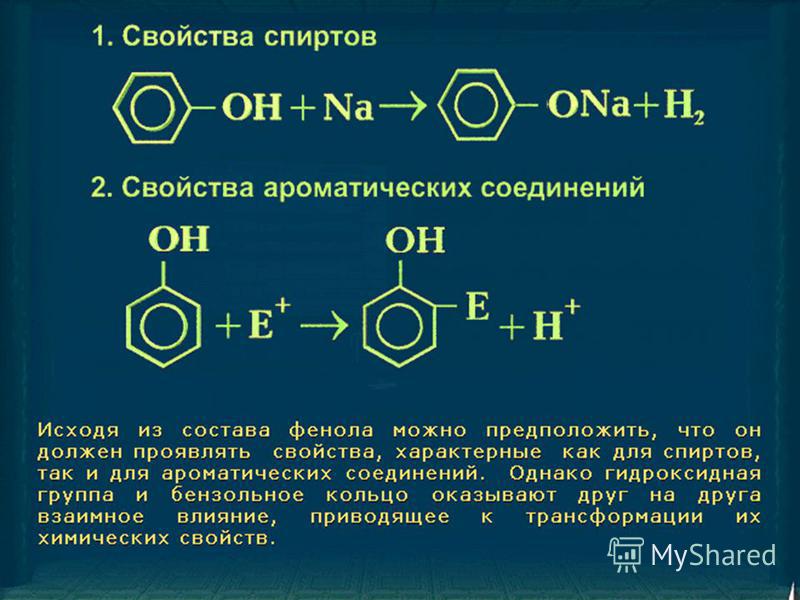
Вы видите, что помимо гидроксильной группы в состав фенолов входит бензольное или ароматическое кольцо, но ни к спиртам, ни к аренам их не относят.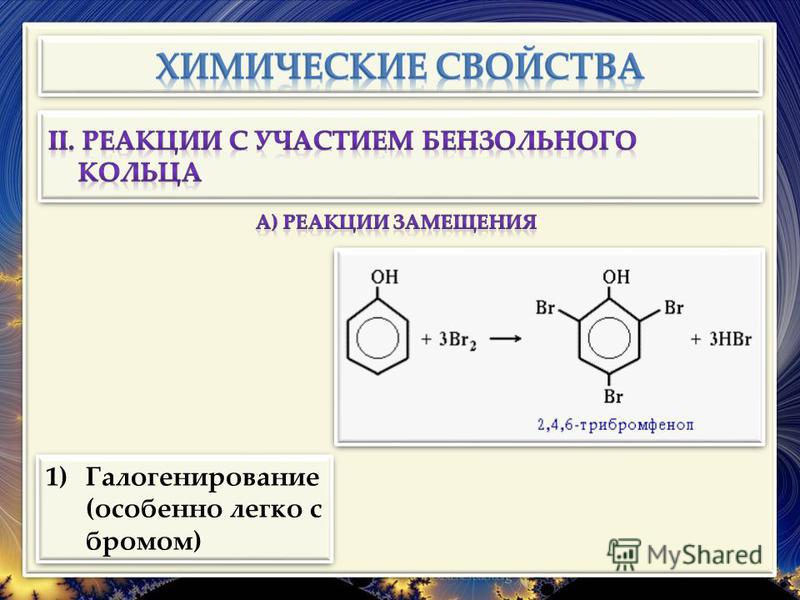 Это отдельный класс органических соединений.
Это отдельный класс органических соединений.
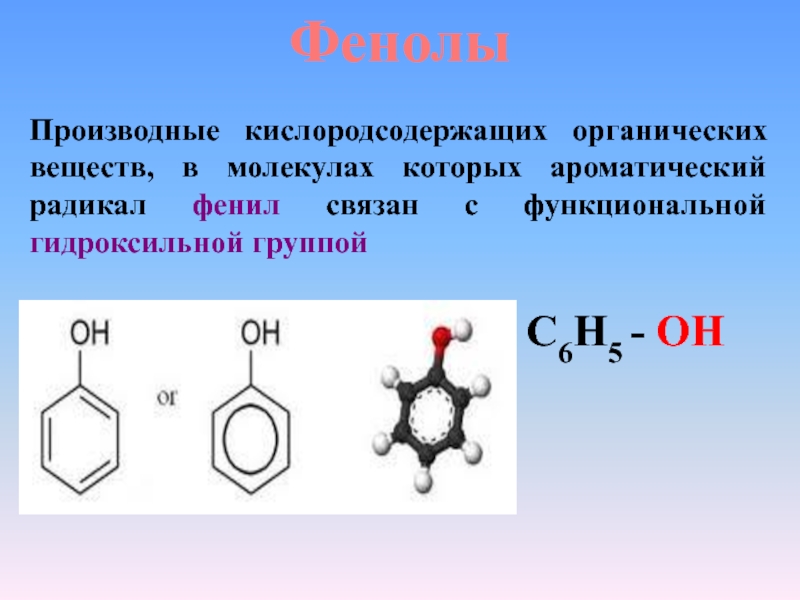
Определение фенолов. Соединения, в которых ароматический радикал фенил С6Н5- непосредственно связан с гидроксильной группой — ОН выделяют в отдельный класс органических соединений, называемый фенолами.
Строение молекулы фенола.
Группа С6Н5- , называемая фенил радикал, обладает свойством притягивать к себе электроны кислородного атома группы –ОН, а значит химическая связь между кислородом и водородом становится более полярной, а атом водорода более подвижный. Это усиливает кислотные свойства фенола.
Но в свою очередь гидроксильная группа –ОН тоже влияет на бензольное кольцо.
Это связано со смещением электронной плотности внутри бензольного кольца. В результате этого химическая связь между С – Н в положениях 2,4 и 6 ослабляется, т.е. атомы водорода в этих положениях становятся более подвижными для реакций замещения, пртекающих по бензольному кольцу.
|
| неподеленная электронная пара атома кислорода притягивается 6-ти электронным облаком бензольного кольца, из – за чего связь О – Н еще сильнее поляризуется. В бензольном кольце нарушается симметричность электронного облака, электронная плотность повышается в положении 2, 4, 6. Это делает более реакционноспособными связи С — Н в положениях 2, 4, 6 и именно в этих положениях проходят реакции замещения. |
 И фенол приобретает кислотные свойства. Фенол- более сильная кислота, чем спирты.
И фенол приобретает кислотные свойства. Фенол- более сильная кислота, чем спирты.Физические свойства фенола. Проведение опыта по растворению фенола
(видеоролик)
Фенол – это бесцветное кристаллическое вещество. При окислении на воздухе кристаллы приобретают розоватый оттенок.
Следствием полярности связи О–Н и наличия неподеленных пар электронов на атоме кислорода является способность гидроксисоединений к образованию водородных связей
Это объясняет, почему у фенола довольно высокие температуры плавления (+43) и кипения (+182). Образование водородных связей с молекулами воды способствует растворимости гидроксисоединений в воде:
Способность растворяться в воде уменьшается с увеличением углеводородного радикала и от многоатомных гидроксисоединений к одноатомным. Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. Растворимость фенола в воде ограничена. Фенол хорошо растворяется в горячей воде. Кроме того, фенол – это ядовитое вещество. При попадании кристалликов фенола на кожу, вызывает сильный химический ожог.
Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. Растворимость фенола в воде ограничена. Фенол хорошо растворяется в горячей воде. Кроме того, фенол – это ядовитое вещество. При попадании кристалликов фенола на кожу, вызывает сильный химический ожог.
Химические свойства фенола (проводится демонстрационный эксперимент)
1) Рассмотрим реакции фенола по ОН- группе: (видеоролик)
Кислотные свойства у фенола выражены сильнее, чем у спирта С2Н5ОН.
Фенол – слабая кислота (карболовая).
2) Реакции фенола по бензольному кольцу: (видеоролик)
Какой вывод о взаимном влиянии атомов в молекуле фенола можно сделать?
Фенильная группа C6H5 – и гидроксил –ОН взаимно влияют друг на друга.
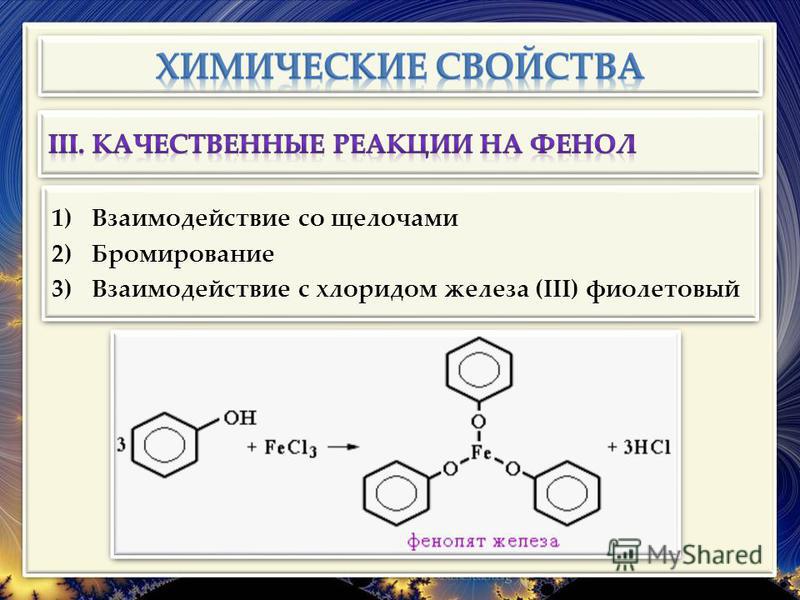
3) Качественная реакция на фенол. Проведение опыта, ( видеоролик)
С6Н5ОН + FeCl3 —> фиолетовое окрашивание
Получение фенола.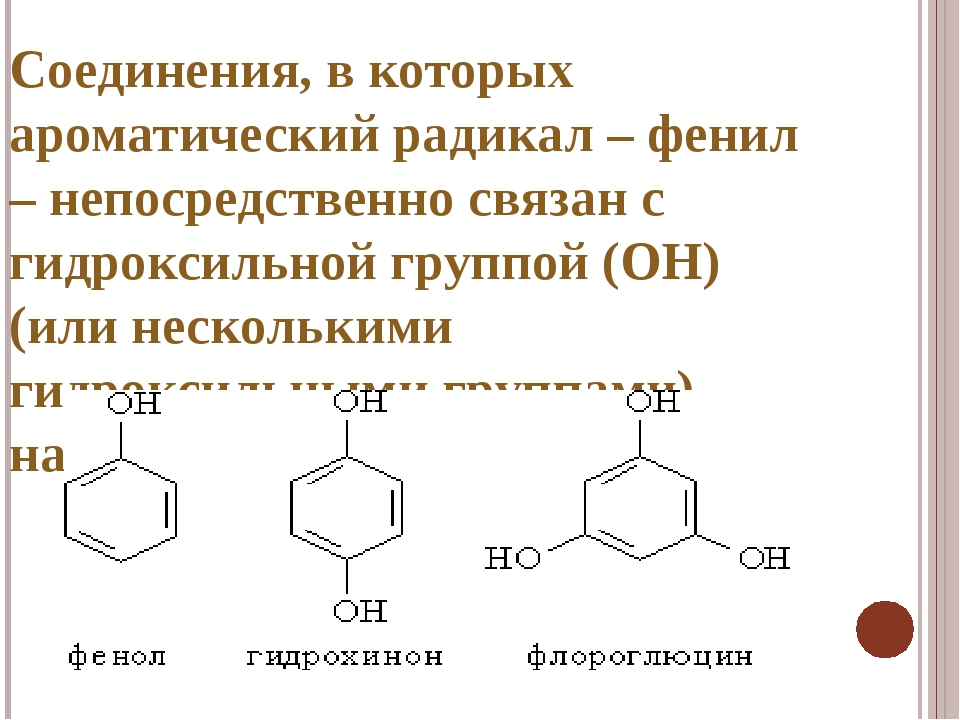
Способы получения фенолов.
Фенолы выделяют из каменноугольной смолы, а также из продуктов пиролиза бурых углей и древесины (деготь).
Промышленный способ получения самого фенола С6Н5ОН основан на окислении ароматического углеводорода кумола (изопропилбензол) кислородом воздуха с последующим разложением получающейся гидроперекиси, разбавленной h3SO4 . Реакция проходит с высоким выходом и привлекательна тем, что позволяет получить сразу два технически ценных продукта – фенол и ацетон.
Другой способ – каталитический гидролиз галогензамещенных бензолов.
Физиологическое действие фенола и его применение
Фенол — ядовит!!! При попадании на кожу вызывает ожоги, при этом он всасывается через кожу и вызывает отравление. Раствор фенола используют в качестве дезинфицирующего средства (карболовая кислота). Двухатомные фенолы – пирокатехин, резорцин, а также гидрохинон (пара-дигидроксибензол) применяют как антисептики (антибактериальные обеззараживающие вещества), вводят в состав дубителей для кожи и меха, как стабилизаторы смазочных масел и резины, а также для обработки фотоматериалов и как реагенты в аналитической химии.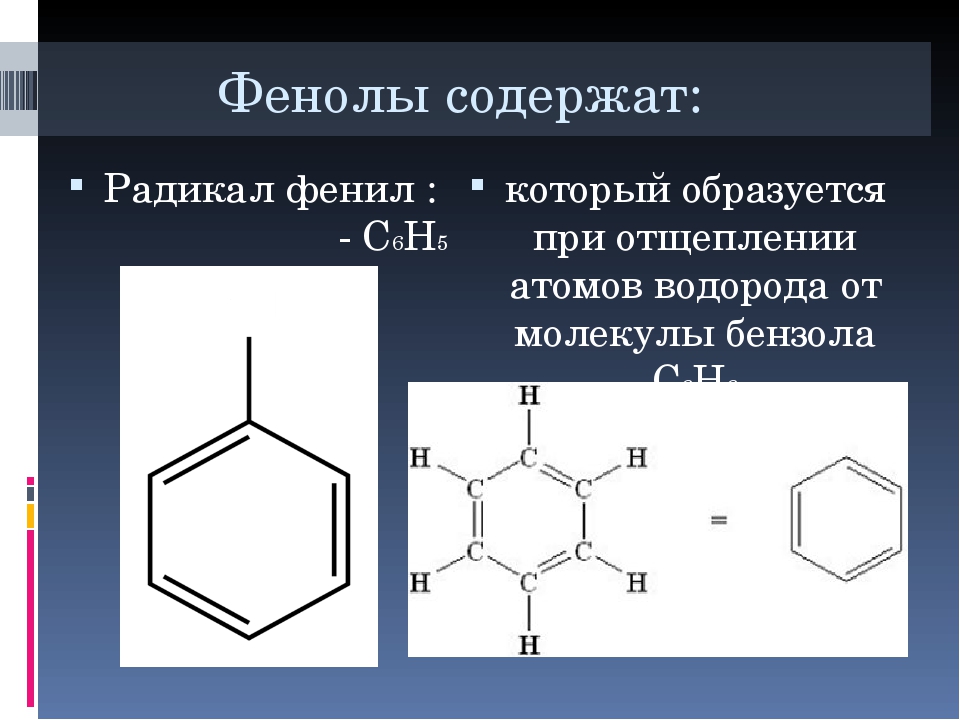
В виде отдельных соединений фенолы используются ограниченно, зато их различные производные применяют широко. Фенолы служат исходными соединениями для получения разнообразных полимерных продуктов – фенолоальдегидных смол, полиамидов, полиэпоксидов.Например: текстолита ( подшипники), карболита (Эл. розетки, вилки), стеклотекстолит, волокнит( электроизоляционные материалы, детали радиоаппаратуры, облицовочные материалы, для получения красителей, лаков, эмалей.
На основе фенолов получают многочисленные лекарственные препараты, например, аспирин, салол, фенолфталеин, кроме того, парфюмерные продукты, пластификаторы для полимеров и средства защиты растений.
Биологическая роль соединений фенола:
| Положительная | Отрицательная (токсическое действие) |
| лекарственные препараты (пурген, парацетамол) антисептики (3-5 % раствор – карболовая кислота) эфирные масла (обладают сильными бактерицидными и противовирусными свойствами, стимулируют иммунную систему, повышают артериальное давление: — анетол в укропе, фенхеле, анисе — карвакрол и тимол в чабреце — эвгенол в гвоздике, базилике Флавоноиды (способствуют удалению радиоактивных элементов из организма) | фенолформальдегидные смолы пестициды, гербициды, инсектициды загрязнение вод фенольными отходами |
4. Закрепление изученного материала.
Закрепление изученного материала.
Какие вещества относят к фенолам?
Как можно распознать фенол среди других веществ?
Какими химическими свойствами обладает фенол?
5. Домашнее задание § 28, стр. 83-85, в. 1.
Фенол радикальные реакции — Справочник химика 21
Поглотительная фракция преимушественно используется для приготовления поглотительного масла для улавливания бензольных углеводородов. Из этой фракции принято извлекать фенолы, так как предполагают, что они снижают стабильность поглотительного масла. Это положение не является бесспорным, так как входящие в состав поглотительной фракции высококипящие фенолы являются ингибиторами радикальных реакций. Поглотительная фракция содержит 4-5% хинолиновых оснований и может явиться источником сырья для производства этих соединений. [c.328]Ингибиторы радикальных реакций — соединения, реагирующие со свободными радикалами, применяются для количественного измерения скорости образования и общего выхода радикалов при распаде пероксидов.
 При этом в первую очередь используют такие соединения, как фенолы, амины, молекулярный иод, стабильные радикалы. В зависимости от химических свойств ингибитора и строения образующихся при распаде радикалов каждая молекула ингибитора реагирует с различным числом радикалов (/). Для акцептирования алкильных радикалов наиболее эффективны стабильные радикалы, иод, хиноны, а для акцептирования окси- и перокси-радикалов — фенолы, ароматические амины. Акцепторы — стабильные радикалы — обычно реагируют с одним радикалом, молекулярные ингибиторы — с двумя. [c.61]
При этом в первую очередь используют такие соединения, как фенолы, амины, молекулярный иод, стабильные радикалы. В зависимости от химических свойств ингибитора и строения образующихся при распаде радикалов каждая молекула ингибитора реагирует с различным числом радикалов (/). Для акцептирования алкильных радикалов наиболее эффективны стабильные радикалы, иод, хиноны, а для акцептирования окси- и перокси-радикалов — фенолы, ароматические амины. Акцепторы — стабильные радикалы — обычно реагируют с одним радикалом, молекулярные ингибиторы — с двумя. [c.61] В механизме ингибирования цепного окисления важную роль играют не только реакции ингибитора (такие, как КОз- + InH), но и реакции образующегося из ингибитора радикала 1п-. В частности, при ингибировании окисления фенолами из них образуются феноксильные радикалы, изучению реакционной способности которых посвящено большое число работ. Наряду с традиционными методами изучения быстрых радикальных реакций (импульсные, струевые) создан ряд методов, где используется специфика ингибированного окисления углеводородов. Эти методы и рассмотрены в настоящем разделе. [c.456]
Эти методы и рассмотрены в настоящем разделе. [c.456]
Окислением алкилароматических углеводородов получают альдегиды, кетоны, кислоты и другие кислородсодержащие соединения. Первичными продуктами при этом будут гидропероксиды. Наибольшее значение в промышленности органического синтеза имеет гидропероксид изопропилбензола, который служит полупродуктом в производстве фенола и ацетона. Его используют в качестве инициатора радикальных реакций полимеризации [c.123]
В этом разделе основное внимание уделено природным антиоксидантам -тем многоатомным фенолам, которые в значительных количествах содержатся в пищевых растениях. Попадая в наш организм с пищей, они проявляют свои ингибирующие свойства в радикальных биохимических процессах. Эта способность фенолов исключительно важна. Как известно, многие формы онкологических заболеваний инициируются активными свободными радикалами. Образуя устойчивые, а потому малореакционноспособные радикалы, многоатомные фенолы обрывают цепи в радикальных реакциях и тем самым тормозят развитие радикальных реакций, в том числе тех, которые сопровождают рост злокачественных опухолей.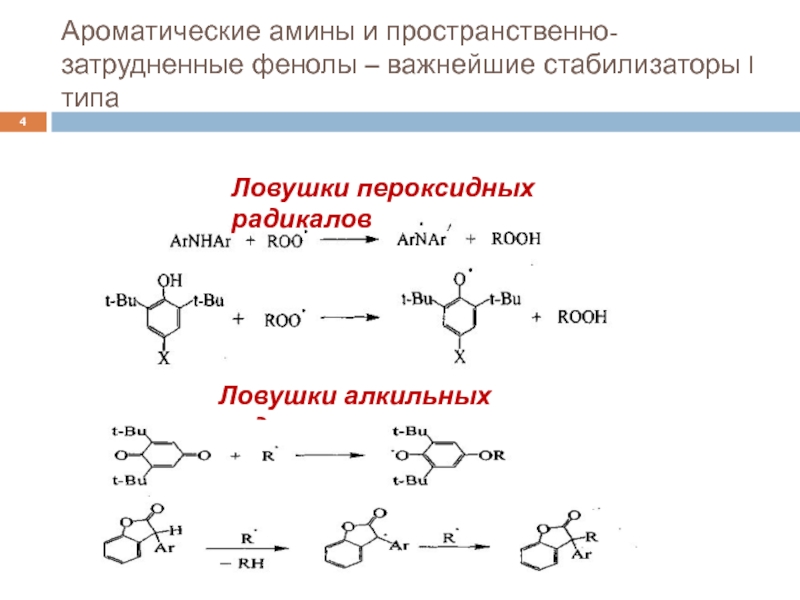 [c.88]
[c.88]
При термическом растворении используются угли крупностью 0,02—0,3 мм, которые смешиваются с растворителем (соотношение от 1 1 до 1 5). В качестве растворителей применяют мазуты, средние и тяжелые масла гидрогенизации, антраценовое масло, дистилляты сланцевых смол, нефтяное дизельное топливо и различные индивидуальные соединения (углеводороды, фенолы, амины и т. д.). Установлено, что хорошие растворители должны обладать дипольным моментом, быть донорами водорода или ингибиторами радикальных реакций. Для крупно-тоннажных производств предпочтительно использовать в качестве растворителей продукты самого процесса термического растворения или легко регенерируемые соединения. При оптимальных условиях проведения процесса и при удачном выборе исходного сырья и растворителя в раствор может переходить до 70—90% органической массы угля. [c.275]
Окисление кумола производится. кислородом воздуха в жидкой фазе в присутствии инициаторов радикальной реакции и марганцевых катализаторов.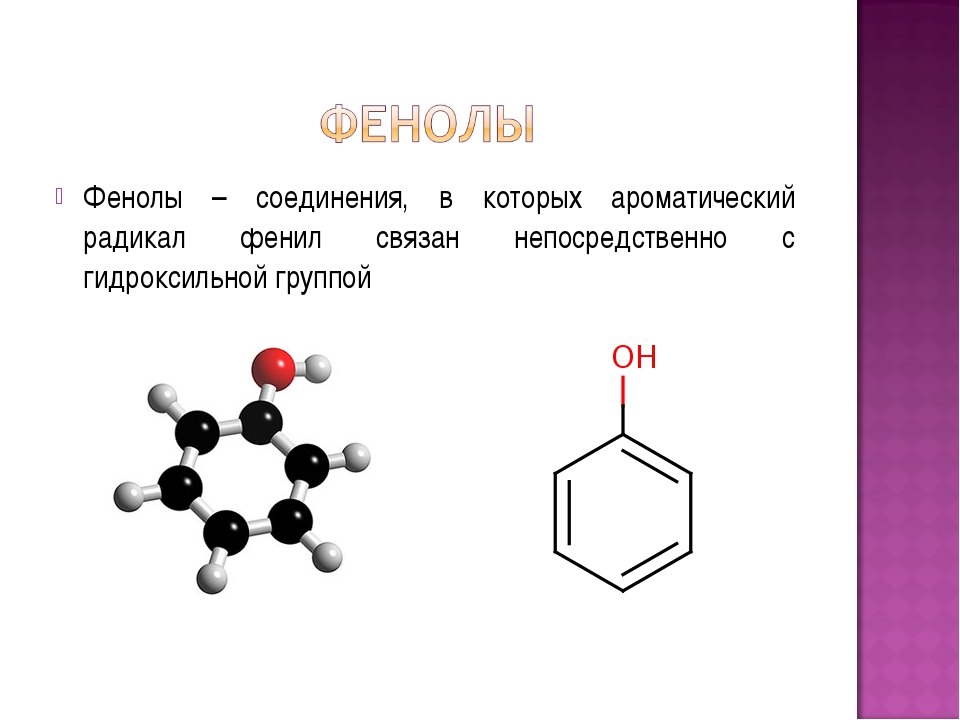 Полученная перекись разлагается на фенол и ацетон в кислой среде (катализаторы — серная кислота, кислый сернистокислый натрий и т. п.). Энергия активации кислотного разложения гидроперекисей составляет, по данным Сергеева 1307], примерно 18.5 ккал/моль. [c.362]
Полученная перекись разлагается на фенол и ацетон в кислой среде (катализаторы — серная кислота, кислый сернистокислый натрий и т. п.). Энергия активации кислотного разложения гидроперекисей составляет, по данным Сергеева 1307], примерно 18.5 ккал/моль. [c.362]
Исследована реакционная способность виниловых эфиров фенола, о-, м-, п-крезолов, п-трет-бутилфенола, м-в п-метоксифенолов, м-ш п-хлорфенолов, и-бром-и и-нитрофенолов, синтезированных на базе ацетилена и фенолов, в реакциях радикальной и ионной полимеризации. [c.416]
Радикальные реакции могут ингибироваться путем введения веществ, которые сами особенно легко реагируют с радикалами (ингибиторы, или улавливатели , радикалов), например фенолов, хинонов, дифениламина, иода и др. Эти и родственные им вещества можно использовать для того, чтобы остановить уже начавшуюся радикальную реакцию. [c.336]
Для физико-химиков основной целью исследований являлось изучение механизма свободно-радикальных реакций фенолов, в частности образования свободных феноксильных радикалов и семихинонных ион-радикалов. Способность фенольных соединений реагировать со свободными радикалами позволила открыть четкие предельные явления в медленных цепных разветвленных реакциях, не говоря уже о том, что в химической кинетике появился новый раздел, посвященный ингибированному окислению. Этот раздел ныне составляет научный фундамент Прикладных исследований в области создания эффективных стабилизаторов полимерных материалов, нефтяных и пищевых продуктов, ингибиторов некоторых биохимических процессов в живых клетках и консервантов для медицинских препаратов. [c.5]
Способность фенольных соединений реагировать со свободными радикалами позволила открыть четкие предельные явления в медленных цепных разветвленных реакциях, не говоря уже о том, что в химической кинетике появился новый раздел, посвященный ингибированному окислению. Этот раздел ныне составляет научный фундамент Прикладных исследований в области создания эффективных стабилизаторов полимерных материалов, нефтяных и пищевых продуктов, ингибиторов некоторых биохимических процессов в живых клетках и консервантов для медицинских препаратов. [c.5]
В 1958—1961 гг. были опубликованы монографии, посвященные торможению процессов окисления пищевых жиров и жиросодержащих продуктов различными антиоксидантами, в том числе фенолами [1, 2] ). Вопросы биохимии катехинов были обобщены в 1964 г. [3]. В 1965 г. особое внимание было уделено обобщению данных по госсиполу — многоатомному природному фенольному соединению [4]. Проведен также анализ результатов но использованию фенолов в качестве ингибиторов свободно-радикальных реакций [5].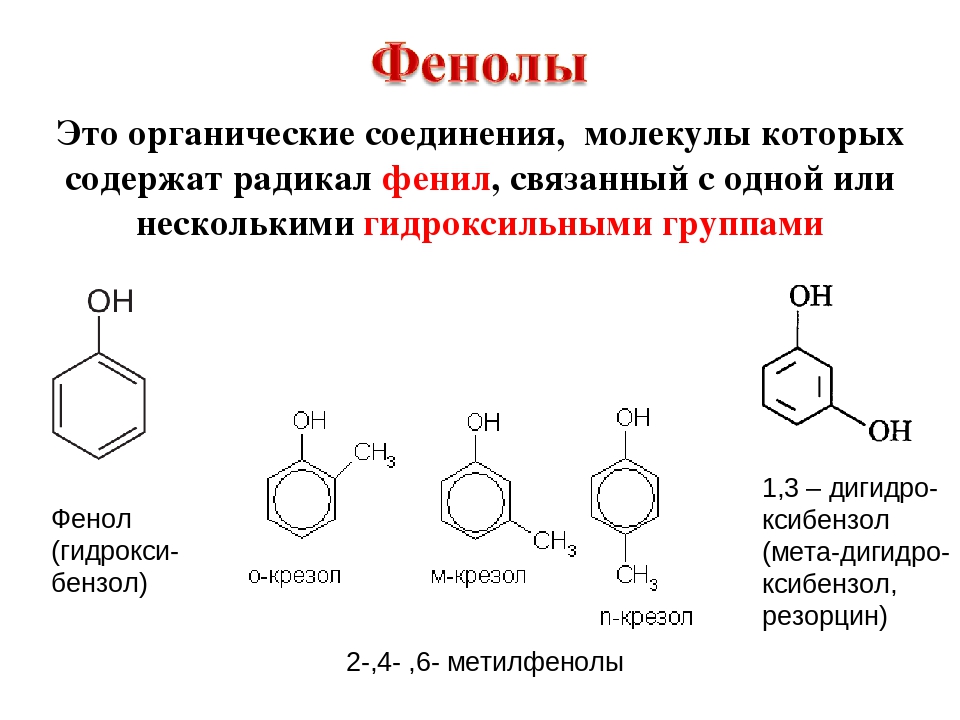 К этому перечню можно отнести также ряд монографий, вышедших за рубежом (например 1.6, 7 ). Кроме того, в самое ближайшее время должен выйти в свет сборник трудов Первого всесоюзного симпозиума по фенольным соединениям, который состоялся в декабре 1966 г. Как видно, список обобщающей литературы не столь уж велик и ни в коей мере не охватывает всего многообразия направлений в изучении фенольных соединений и их интересных и ценных свойств. Нужно ли после этого говорить, сколь важным было появление в свет сборника под редакцией Харборна, изданного в 1964 г. и посвященного биохимии фенольных соединений, который сейчас предлагается читателям в русском переводе. [c.5]
К этому перечню можно отнести также ряд монографий, вышедших за рубежом (например 1.6, 7 ). Кроме того, в самое ближайшее время должен выйти в свет сборник трудов Первого всесоюзного симпозиума по фенольным соединениям, который состоялся в декабре 1966 г. Как видно, список обобщающей литературы не столь уж велик и ни в коей мере не охватывает всего многообразия направлений в изучении фенольных соединений и их интересных и ценных свойств. Нужно ли после этого говорить, сколь важным было появление в свет сборника под редакцией Харборна, изданного в 1964 г. и посвященного биохимии фенольных соединений, который сейчас предлагается читателям в русском переводе. [c.5]
В настоящее время накопилась обширная литература, затрагивающая различные вопросы химии пространственно-затрудненных фенолов, однако каких-либо обзорных работ в этой области нет. В предлагаемой книге авторы обобщили большую литературу по химии и применению пространственно-затрудненных фенолов. Работая в этой области, авторы критически, на основе современных теоретических представлений переработали литературный материал и внесли много нового для понимания сущности процессов, протекающих с участием пространственно-затрудненных фенолов. Большое внимание в книге уделено строению и связанным с ним физическим свойствам пространственно-затрудненных фенолов, реакциям электрофильного замещения и радикальным реакциям. В последних главах авторы приводят фактический материал по практическому применению пространственно-затрудненных фенолов и их производных. В целом в книге удачно сочетаются общие и теоретические вопросы химии пространственно-затрудненных фенолов с проблемами прикладного характера. [c.6]
Большое внимание в книге уделено строению и связанным с ним физическим свойствам пространственно-затрудненных фенолов, реакциям электрофильного замещения и радикальным реакциям. В последних главах авторы приводят фактический материал по практическому применению пространственно-затрудненных фенолов и их производных. В целом в книге удачно сочетаются общие и теоретические вопросы химии пространственно-затрудненных фенолов с проблемами прикладного характера. [c.6]
Участие пространственно-затрудненных фенолов в радикальных реакциях в общем виде сводится к первоначальному отрыву атома водорода от гидроксильной группы фенола с образованием фен-оксильного радикала, дальнейшая судьба которого зависит от его реакционной способности, присутствия кислорода, реакционной среды, температуры и концентрации реагирующих веществ. Другими словами, поведение пространственно-затрудненных фенолов в радикальных реакциях определяется свойствами образующихся из них феноксильных радикалов.
 Поэтому в настоящей главе основное внимание уделено рассмотрению химических и физических свойств феноксильных радикалов. [c.90]
Поэтому в настоящей главе основное внимание уделено рассмотрению химических и физических свойств феноксильных радикалов. [c.90]При окислении пространственно-затрудненных фенолов кислородом первоначально также образуется соответствующий фенок-сильный радикал. Следовательно, окисление пространственно-затрудненных фенолов кислородом представляет собой типичную радикальную реакцию, подчиняющуюся общим закономерностям рассмотренных выше радикальных процессов. [c.157]
Для ингибирования радикальных процессов применяются пространственно затрудненные фенолы, ароматические амины, хиноны и некоторые другие соединения. Ингибиторы в результате взаимодействия с активными свободными радикалами образуют малоактивные ргдикалы, не способные продолжать развитие цепной радикальной реакции. Ниже в качестве примера приведены схемы взаимодействия пероксидных радикалов с ионолом и дифениламином [c.148]
Основными отличительными чертами радикальных реакций являются высокая скорость, способность инициироваться радикалами или образующими их веществами (инициаторами), а также способность замедляться или полностью превращаться в присутствии веществ, активно реагирующих с радикалами (ингибиторов), таких, как, например, фенолы, хиноны, дифениламин, иод и др.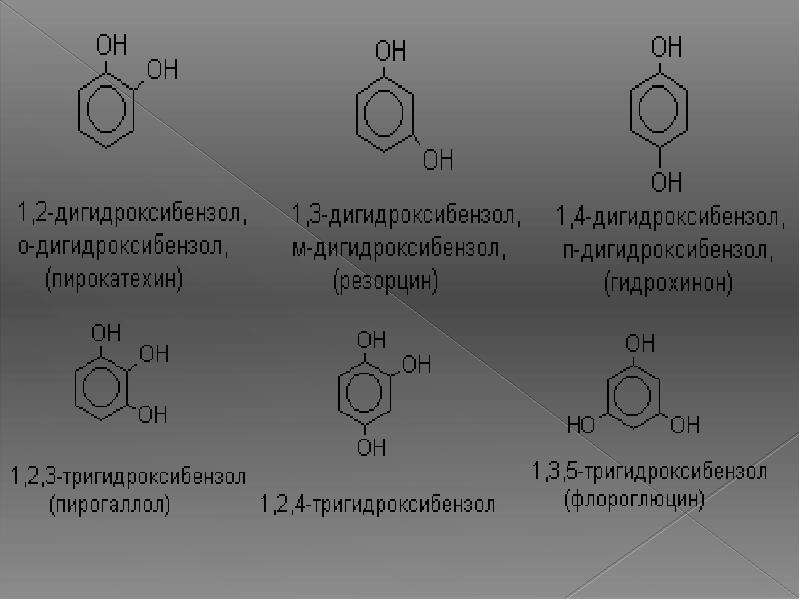 Помимо неустойчивых короткоживущих радикалов, которые возникают в реакции лищь как промежуточные продукты, известен ряд устойчивых радикалов, способных существовать в течение продолжительного времени с них мы и начнем рассмотрение. [c.277]
Помимо неустойчивых короткоживущих радикалов, которые возникают в реакции лищь как промежуточные продукты, известен ряд устойчивых радикалов, способных существовать в течение продолжительного времени с них мы и начнем рассмотрение. [c.277]
Способность ДФПГ взаимодействовать с радикалами позволяет использовать его в качестве ингибитора таких радикальных реакций, как полимеризация и радикальное цепное окисление. ДФПГ используют в качестве одноэлектронного окислителя для окисления тиолов и фенолов. В качестве основного продукта окисления тиолов образуется дисульфид [c.520]
Хемилюминесцентным методом в неводных растворах предложено [39] определять ингибиторы свободно-радикальных реакций окисления ряда углеводородов с чувствительностью — 10″ М. (р-нафтол, неозон А, а-наф-тол, тетраизопропилфенолфталеин, дифениламин, фенол). В качестве параметра измерений выбрана начальная интенсивность и время, необходимое для достижения определенной интенсивности свечения. [c.85]
[c.85]
Присутствие некоторых загрязнений оказывает большое влияние на реакцию окисления с образованием гидроперекисей аналогично тому, что имеет место при свободно-радикальной полимеризации. Фенолы, например, при концентрациях порядка 10 моля на 1 моль окисляюш,егося веш,ества почти полностью подавляют реакцию [24, 251. Соотношения между различными процессами, по которым могут реагировать радикалы, принимаюш,ие участие в цепной реакции, подробно рассмотрены для случая полимеризации [61, 62], причем было проведено различие между ингибированием и замедлением . Реакция ингибируется, когда введенная примесь реагирует с инициирующими радикалами раньше, чем они смогут развить цепи, причем этот процесс настолько эффективен, что в идеальном случае цепная реакция полностью подавляется до тех пор, пока не израсходуется весь ингибитор. Начиная с этого момента цепная реакция протекает с такой же скоростью, как и в отсутствие ингибитора, причем время полного ингибирования пропорционально концентрации ингибитора. Замедление же наблюдается в тех случаях, когда обрыв цепей в результате взаимодействия радикалов с молекулами примеси конкурирует с реакцией обрыва, протекающей по обычному механизму. Таким образом, замедлитель только несколько снижает скорость реакции. По мере израсходования замедлителя скорость реакции постепенно увеличивается до значения, наблюдаемого в отсутствие замедлителя. Согласно этим определениям, влияние фенолов на реакции окисления можно назвать замедлением. [c.148]
Замедление же наблюдается в тех случаях, когда обрыв цепей в результате взаимодействия радикалов с молекулами примеси конкурирует с реакцией обрыва, протекающей по обычному механизму. Таким образом, замедлитель только несколько снижает скорость реакции. По мере израсходования замедлителя скорость реакции постепенно увеличивается до значения, наблюдаемого в отсутствие замедлителя. Согласно этим определениям, влияние фенолов на реакции окисления можно назвать замедлением. [c.148]
При сопоставлении логарифмов констант Л ингибиторов с константами а Гамметта [13] заместителей обнаружена хорошая линейная корреляция (рис. 4), позволяюш ая говорить о том, что скорость радикальных реакций отрыва водорода от ОН-группы в феноле пероксирадикалом зависит от влияния полярности заместителя, а это является следствием разной электронной плотности в реакционном центре фенола — гидроксильной группе [17]. Для незамещенных диоксибензолов рассчитана электронная плотность на атоме кислорода м — 0,087, о — 0,078, п — 0,075) [16] и установлено, что она уменьшается в ряду л- > о- > п-.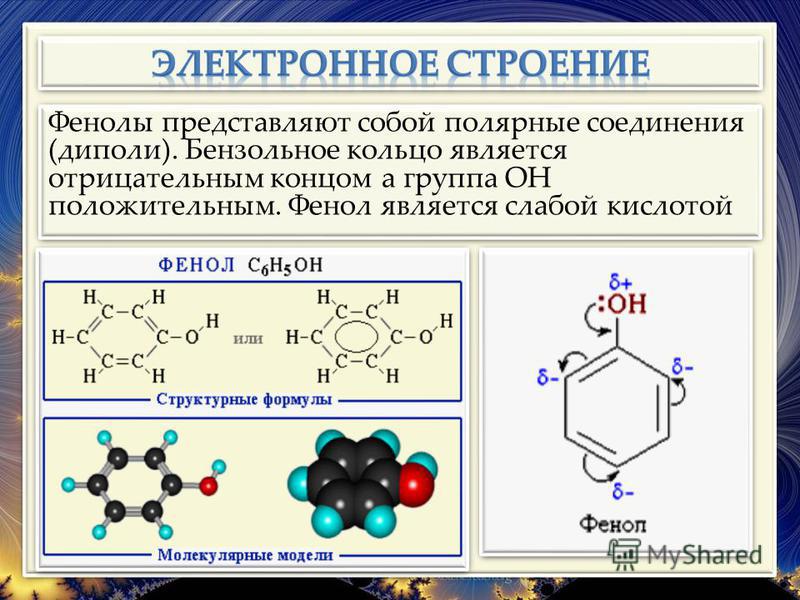 Эти данные находятся в согласии с экспериментальными данньгаи по ингибирующей способности диоксибензолов. [c.243]
Эти данные находятся в согласии с экспериментальными данньгаи по ингибирующей способности диоксибензолов. [c.243]
Из этой таблицы видно, что фенол можно получить с выходом от 50 до 80%, однако только в том случае, если конверсия бензола будет 10% или ниже. В своей работе Мойер [129] указывает, что гомогенное парофазное окисление бензола в фенол тормозится при помещении инертной насадки в реактор. Однако, если инертную насадку покрыть ВгОз, то превращения, выраженные в процентах, будут равны превращениям в реакторе без насадки. Вероятно, В2О3 предотвращает обрыв цепи свободно-радикальной реакции па поверхности, что облегчает гомогенное окисление. [c.216]
Миграция ацильной группы не обязательно связана с ионными или радикальными реакциями, но может быть результатом сигма-тропных сдвигов в диеноновых интермедиатах, известных в химии фенолов. Так, в результате перегруппировки Кляйзена аллилового [c.249]
Назначение антиокислителя заключается в подавлении или полном устранении стадий распространения и разветвления цепи.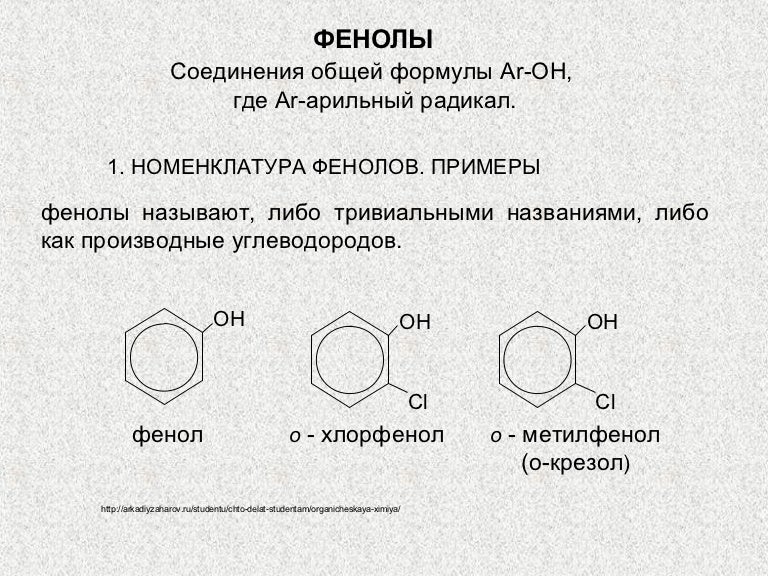 Распространение цепи можно затормозить добавлением соединений, легко отдающих водород или связываюших перекисные радикалы (фенолы и ароматические амины). Разветвление цепи можно подавить добавлением продуктов, взаи-модействуюших с гидроперекисью (антиокислители типа сульфидов и алифатических аминов). В качестве присадок, подавляюших образование перекисных цепей и радикальные реакции других реакционноспособных компонентов топлива, например образование тиильных радикалов из активных меркаптанов, применяются некоторые арилалкиламины. [c.313]
Распространение цепи можно затормозить добавлением соединений, легко отдающих водород или связываюших перекисные радикалы (фенолы и ароматические амины). Разветвление цепи можно подавить добавлением продуктов, взаи-модействуюших с гидроперекисью (антиокислители типа сульфидов и алифатических аминов). В качестве присадок, подавляюших образование перекисных цепей и радикальные реакции других реакционноспособных компонентов топлива, например образование тиильных радикалов из активных меркаптанов, применяются некоторые арилалкиламины. [c.313]
При механич. воздействии на П. (перетирании, вальцевании и др.) образуются макрорадикалы, рекомбинация к-рых приводит к синтезу блоксополимеров, а при протекании реакции передачи цепи — привитых сополимеров и интерполимеров (механохимич. метод получения привитых и блоксополимеров). Если механич. разрыв макромолекул происходит в среде мономера, то возникающие макрорадикалы инициируют полимеризацию этого мономера. Эффективность механодеструкции П.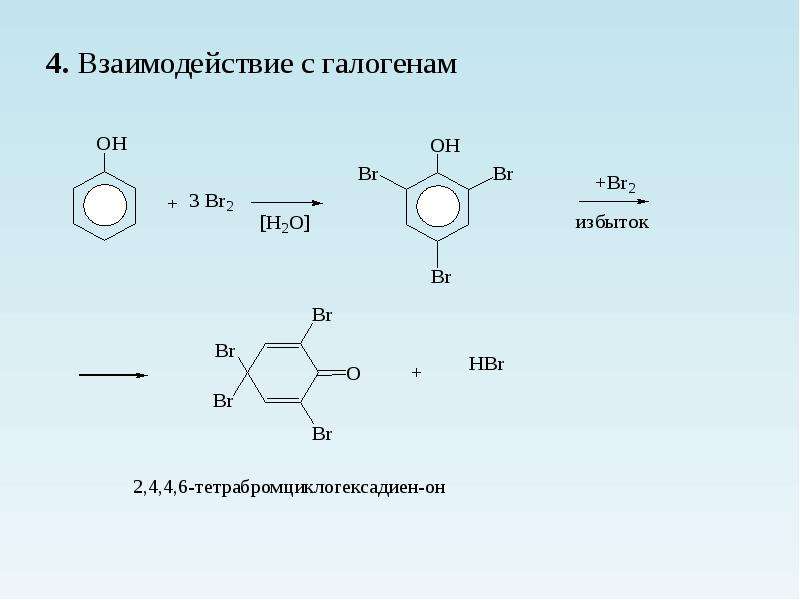 возрастает при понижении темп-ры, особенно ниже темп-ры стеклования (70—80 °С). Процесс ингибируется кислородом и присутствующими в зоне реакции ингибиторами радикальных реакций. Получены смеси привитых сополимеров, блоксополимеров и интерполимеров поливинилхлорида с новолачными феноло-формальдегидными смолами, полиметилметакрилатом и полистиролом (вальцевание), с хлоронреновым каучуком (экструзия). При пластикации поливинилхлорида в смеси с малеиновым ангидридом и др. мономерами, а также при вибропомоле полиметилметакрилата или полиакрилонитрила с В. получены только привитые сополимеры, а при использовании электрогидравлич. эффекта (импульсы давления, возникающие при высоковольтных искровых разрядах в р-ре полимера) — привитые и блоксополимеры, напр, поливинилхлорида с метилметакрилатом или этилцеллюлозой (в р-ре циклогексанона). [c.226]
возрастает при понижении темп-ры, особенно ниже темп-ры стеклования (70—80 °С). Процесс ингибируется кислородом и присутствующими в зоне реакции ингибиторами радикальных реакций. Получены смеси привитых сополимеров, блоксополимеров и интерполимеров поливинилхлорида с новолачными феноло-формальдегидными смолами, полиметилметакрилатом и полистиролом (вальцевание), с хлоронреновым каучуком (экструзия). При пластикации поливинилхлорида в смеси с малеиновым ангидридом и др. мономерами, а также при вибропомоле полиметилметакрилата или полиакрилонитрила с В. получены только привитые сополимеры, а при использовании электрогидравлич. эффекта (импульсы давления, возникающие при высоковольтных искровых разрядах в р-ре полимера) — привитые и блоксополимеры, напр, поливинилхлорида с метилметакрилатом или этилцеллюлозой (в р-ре циклогексанона). [c.226]
Способность фенолов задерживать и инициировать радикальные реакции объясняет многообразие их свойств. При воздействии ПГ была установлена потеря бластомогенных свойств вируса саркомы Рауса [69], Обнаружено подавление пропилгаллатом вируса ядер-ного полиэдроза дубового шелкопряда [70]. При этом Кок [71 показала, что под действием ПГ в тканях куг олок шелкопряда уменьшается количество нуклеиновых кислот. Известно, что фенольные ооединения подавляют развитие растительных раков. Так, ПГ подавляет развитие рака картофеля, вызванного Ba terium iumifd iens [72], Подобный эффект был получен на опухолях подсолнечника и томатов, вызванных теми же бактериями [73], [c.328]
При этом Кок [71 показала, что под действием ПГ в тканях куг олок шелкопряда уменьшается количество нуклеиновых кислот. Известно, что фенольные ооединения подавляют развитие растительных раков. Так, ПГ подавляет развитие рака картофеля, вызванного Ba terium iumifd iens [72], Подобный эффект был получен на опухолях подсолнечника и томатов, вызванных теми же бактериями [73], [c.328]
Радикальные реакции пространственно-затрудненных фенолов стали широко исследоваться в связи с тем, что соединения этого ряда оказались весьма эффективными ингибиторами окислительных процессов различных органических вешеств. Первичные продукты радикальных реакций пространственно-затрудненных фенолов— феноксильные радикалы — представляют и самостоятельный интерес, так как могут служить удобной моделью для изучения механизма различных процессов, протекающих с образованием в качестве промежуточных продуктов свободных радикалов. Мощным стимулом в развитии химии феноксильных радикалов явилось создание и усовершенствованиие метода ЭПР, который позволил исследовать не только структуру радикалов, но и кинетику радикальных процессов. [c.90]
[c.90]
Все указанные методы применимы и для получения стабильных феноксильных радикалов—ароксилов. Физические свойства и химические превращения ароксилов широко представлены в химической литературе Поэтому в настоящей главе основное внимание уделено связи реакционной способности феноксильных радикалов с их электронным строением, влиянию стерических эффектов орто-заместителей на направление радикальных реакций, а также собственно радикальным реакциям пространственно-затрудненных фенолов и феноксильных радикалов. Бирадикалы кар-бенового типа рассматриваются в разделе химии п-бензохинонди-азидов (см. гл. 9)., [c.91]
Таким образом, приведенные выше факты показывают, что благодаря взаимосвязи зарядовой и спиновой плотностей неспаренного электрона последняя может служить критерием реакционной способности феноксильных радикалов. Это позволяет предвидеть возможные напра вления радикальных реакций, предсказывать строение и соотношение конечных продуктов.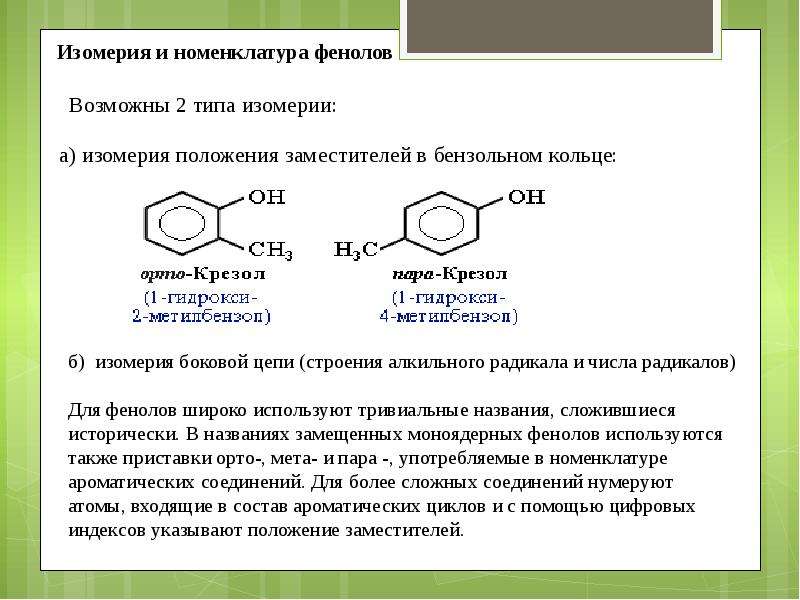 Естественно, что при этом необходимо учитывать полярные и стерические эффекты заместителей, которые часто вызывают аномальное, на первый взгляд, течение реакций. Большую информацию могут дать результаты, полученные при использовании корреляционных уравнений, отражающих влияние полярных факторов на реакционную способность молекулой радикалов. Так, при исследовании реакции а,а-дифенил-пикрилгидразила с различными фенолами было показаночто с увеличением электроноакцепторности заместителей в феноле константа скорости гомолитического отрыва атома водорода от гидроксильной группы фенола уменьшается. Аналогичное исследование было проведено и на примере взаимодействия 2,4,6-три-т рег-бутилфеноксила с пара-замещенными фенолами [c.115]
Естественно, что при этом необходимо учитывать полярные и стерические эффекты заместителей, которые часто вызывают аномальное, на первый взгляд, течение реакций. Большую информацию могут дать результаты, полученные при использовании корреляционных уравнений, отражающих влияние полярных факторов на реакционную способность молекулой радикалов. Так, при исследовании реакции а,а-дифенил-пикрилгидразила с различными фенолами было показаночто с увеличением электроноакцепторности заместителей в феноле константа скорости гомолитического отрыва атома водорода от гидроксильной группы фенола уменьшается. Аналогичное исследование было проведено и на примере взаимодействия 2,4,6-три-т рег-бутилфеноксила с пара-замещенными фенолами [c.115]
Методы получения метиленхйнонов. Образование метиленхинонов происходит при многих радикальных реакциях пространственно-затрудненных фенолов (см. гл. 5). Наиболее общим методом получения 2,6-диалкилметиленхинонов является окисление пространственно-затрудненных фенолов соединениями металлов переменной валентности феррицианидом калия в щелочной сре-Д0 85, 93, 98,99 двуокисью свинца ОКИСЬЮ Серебра [c.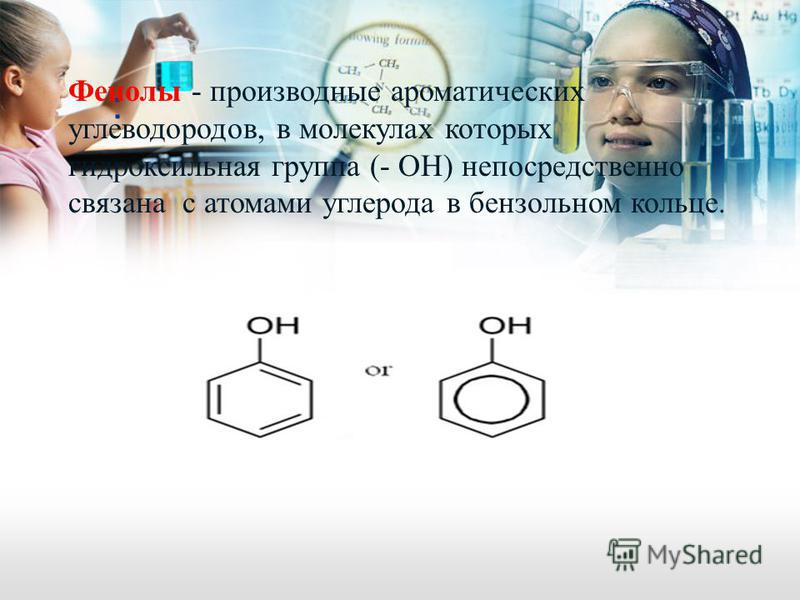 247]
247]
Молекулярная и структурная формула расчета фенола
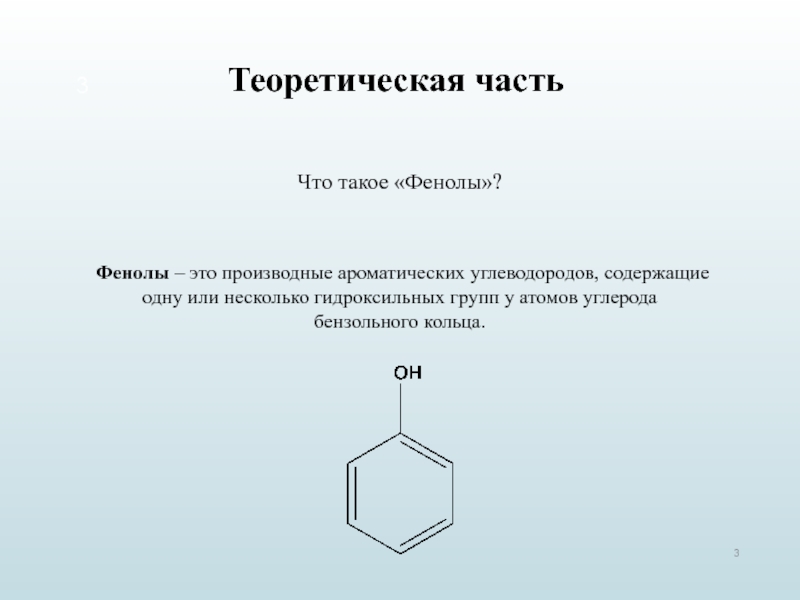
Фенолы — общее название ароматических спиртов. По своим свойствам вещества являются слабыми кислотами. Важное практическое значение имеют многие гомологи гидроксибензола С6Н50Н (формула фенола) — простейшего представителя класса. Остановимся на этом подробнее.
Фенолы. Общая формула и классификация
Общая формула органических веществ, относящихся к ароматическим спиртам, — R-OH. Молекулы собственно фенолов и крезолов образованы радикалом — фенилом С6Н5, с которым непосредственно соединяется одна или несколько гидроксильных групп OH (оксигрупп). По их числу в молекуле фенолы классифицируются на одно-, двух- и многоатомные. Одноатомными соединениями этого типа являются фенол и крезол. Наиболее распространенные среди многоатомных гидроксибензолов — нафтолы, которые содержат в своем составе 2 конденсированных ядра.
Фенол — представитель ароматических спиртов
Текстильщикам фенол был известен уже в XVIII веке: ткачи использовали его в качестве красителя. При перегонке каменноугольной смолы в 1834 году химик из Германии Ф. Рунге выделил кристаллы этого вещества с характерным сладковатым запахом. Латинское название угля — carbo, поэтому соединение называли карболовой кислотой (карболкой). Немецкому исследователю не удалось определить состав вещества. Молекулярная формула фенола была установлена в 1842 годах О. Лораном, считавшим карболку производным бензола. Для новой кислоты употребляли наименование «фениловая». Шарль Жерар определил, что вещество является спиртом, и назвал его фенолом. Первоначальные области применения соединения — медицина, дубление кож, выпуск синтетических красителей. Характеристики рассматриваемого вещества:
При перегонке каменноугольной смолы в 1834 году химик из Германии Ф. Рунге выделил кристаллы этого вещества с характерным сладковатым запахом. Латинское название угля — carbo, поэтому соединение называли карболовой кислотой (карболкой). Немецкому исследователю не удалось определить состав вещества. Молекулярная формула фенола была установлена в 1842 годах О. Лораном, считавшим карболку производным бензола. Для новой кислоты употребляли наименование «фениловая». Шарль Жерар определил, что вещество является спиртом, и назвал его фенолом. Первоначальные области применения соединения — медицина, дубление кож, выпуск синтетических красителей. Характеристики рассматриваемого вещества:
- Рациональная химическая формула — C6H5OH.
- Молекулярная масса соединения — 94,11 а. е. м.
- Брутто-формула, отражающая состав, — C6H6O.
Электронное и пространственное строение молекулы фенола
Циклическую структурную формулу бензола предложил немецкий химик-органик Ф. Кекуле в 1865 году, а незадолго до него — И. Лошмидт. Ученые представляли молекулу органического вещества в виде правильного шестиугольника с чередующимися простыми и двойными связями. По современным представлениям, ароматическое ядро — это особый вид кольцевой структуры, получивший название «сопряженная связь».
Кекуле в 1865 году, а незадолго до него — И. Лошмидт. Ученые представляли молекулу органического вещества в виде правильного шестиугольника с чередующимися простыми и двойными связями. По современным представлениям, ароматическое ядро — это особый вид кольцевой структуры, получивший название «сопряженная связь».
Шесть атомов углерода С испытывают процесс sp2-гибридизации электронных орбиталей. Не участвующие в образовании С—С-связей р-электронные облака перекрываются над и под плоскостью ядра молекулы. Возникают две части общего электронного облака, которое охватывает все кольцо. Структурная формула фенола может выглядеть по-разному, учитывая исторический подход к описанию строения бензола. Чтобы подчеркнуть непредельный характер ароматических углеводородов, условно считают двойными три из шести связей, которые перемежаются с тремя простыми.
Поляризация связи в оксигруппе
В простейшем ароматическом углеводороде — бензоле С6Н6 — электронное облако является симметричным. Формула фенола отличается на одну оксигруппу. Присутствие гидроксила нарушает симметрию, что находит отражение в свойствах вещества. Связь между кислородом и водородом в оксигруппе — полярная ковалентная. Смещение общей пары электронов к атому кислорода приводит к возникновению на нем отрицательного заряда (частичного). Водород лишается электрона и приобретает частичный заряд «+». Кроме того, кислород в О—Н-группе является обладателем двух неподеленных электронных пар. Одна из них притягивается электронным облаком ароматического ядра. По этой причине связь становится более поляризованной, атом водорода легче замещается металлами. Модели дают представления о несимметричном характере молекулы фенола.
Формула фенола отличается на одну оксигруппу. Присутствие гидроксила нарушает симметрию, что находит отражение в свойствах вещества. Связь между кислородом и водородом в оксигруппе — полярная ковалентная. Смещение общей пары электронов к атому кислорода приводит к возникновению на нем отрицательного заряда (частичного). Водород лишается электрона и приобретает частичный заряд «+». Кроме того, кислород в О—Н-группе является обладателем двух неподеленных электронных пар. Одна из них притягивается электронным облаком ароматического ядра. По этой причине связь становится более поляризованной, атом водорода легче замещается металлами. Модели дают представления о несимметричном характере молекулы фенола.
Особенности взаимовлияния атомов в феноле
Единое электронное облако ароматического ядра в молекуле фенола взаимодействует с гидроксильной группой. Происходит явление, получившее название сопряжения, в результате которого собственная пара электронов атома кислорода оксигруппы притягивается к системе бензольного цикла.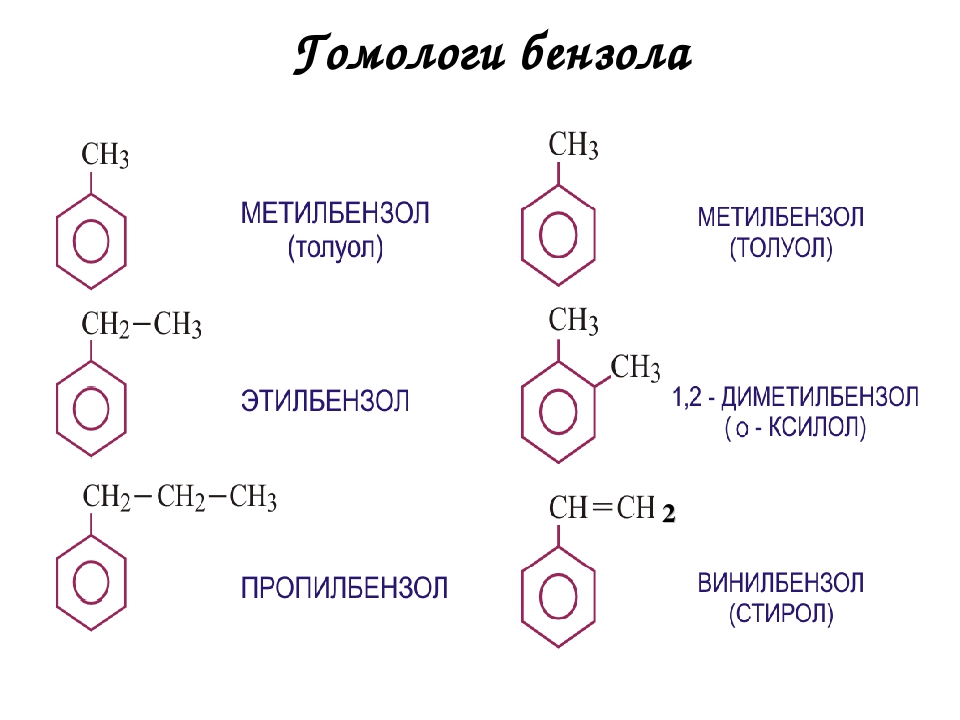 Снижение отрицательного заряда компенсируется благодаря еще большей поляризации связи в группе О—Н.
Снижение отрицательного заряда компенсируется благодаря еще большей поляризации связи в группе О—Н.
В ароматическом ядре также изменяется система электронного распределения. Она понижается на углероде, который связан с кислородом, и повышается у ближайших к нему атомов, находящихся в орто-положениях (2 и 6). Сопряжение вызывает накопление на них заряда «–». Дальнейшее» смещение плотности — движение ее от атомов в мета-положениях (3 и 5) к углероду в пара-положении (4). Формула фенола для удобства изучения сопряжения и взаимовлияния обычно содержит нумерацию атомов бензольного кольца.
Объяснение химических свойств фенола на основе его электронного строения
Процессы сопряжения ароматического ядра и гидроксила сказываются на свойствах обеих частиц и всего вещества. Например, высокая электронная плотность у атомов в орто- и пара-положениях (2, 4, 6) делает С—Н-связи ароматического цикла фенола более реакционноспособными. Снижается отрицательный заряд атомов углерода в мета-положениях (3 и 5).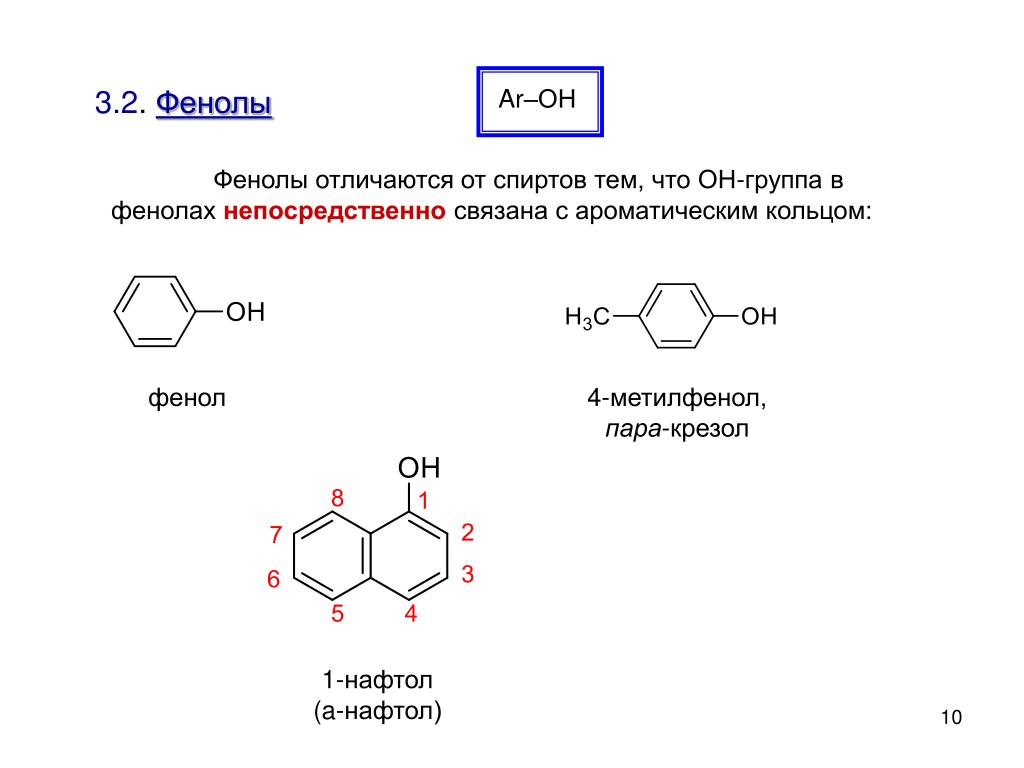 Атаке электрофильных частиц в химических реакциях подвергается углерод, находящийся в орто- и пара-положениях. В реакции бромирования бензола изменения наступают при сильном нагревании и присутствии катализатора. Образуется моногалогенопроизводное — бромбензол. Формула фенола позволяет веществу реагировать с бромом практически мгновенно без нагревания смеси.
Атаке электрофильных частиц в химических реакциях подвергается углерод, находящийся в орто- и пара-положениях. В реакции бромирования бензола изменения наступают при сильном нагревании и присутствии катализатора. Образуется моногалогенопроизводное — бромбензол. Формула фенола позволяет веществу реагировать с бромом практически мгновенно без нагревания смеси.
Ароматическое ядро влияет на полярность связи в оксигруппе, увеличивая ее. Атом водорода становится подвижнее, по сравнению с предельными спиртами. Фенол реагирует со щелочами, образуя соли – феноляты. Этанол не взаимодействует со щелочами, вернее, продукты реакции — этаноляты — разлагаются. В химическом плане фенолы — более сильные кислоты, чем спирты.
Представители класса ароматических спиртов
Брутто-формула гомолога фенола — крезола (метилфенола, гидрокситолуола) — C7H8O. Вещество в природном сырье часто сопутствует фенолу, тоже обладает антисептическими свойствами. Другие гомологи фенола:
- Пирокатехин (1,2-гидроксибензол).
 Химическая формула — С6Н4(ОН)2.
Химическая формула — С6Н4(ОН)2. - Резорцин (1,3-гидроксибензол) — С6Н4(ОН)2.
- Пирогаллол (1,2,3- тригидроксибензол) — С6Н3(ОН)3.
- Нафтол. Брутто-формула вещества — C10H7OH. Применяется в производстве красителей, медикаментов, душистых соединений.
- Тимол (2-изопропил-5-метилфенол). Химическая формула — C6H3CH3(OH)(C3H7). Применяется в химии органического синтеза, медицине.
- Ванилин, кроме фенольного радикала, содержит простую эфирную группу и остаток альдегида. Брутто-формула соединения — C8H8O3. Ванилин широко используется как искусственная отдушка.
Формула реактива для распознавания фенолов
Качественное определение фенола можно проводить с помощью брома. В результате реакции замещения выпадает белый осадок трибромфенола. Пирокатехин (1,2-гидроксибензол) окрашивается в зеленый цвет в присутствии растворенного хлорида трехвалентного железа. С этим же реагентом вступает в химическую реакцию фенол, и образуется трифенолят, обладающий фиолетовым цветом. Качественная реакция на резорцин — появление темно-фиолетового окрашивания в присутствии хлорида трехвалентного железа. Постепенно цвет раствора становится черным. Формула реактива, который служит для распознавания фенола и некоторых его гомологов, — FeCl3 (хлорид железа (III)).
С этим же реагентом вступает в химическую реакцию фенол, и образуется трифенолят, обладающий фиолетовым цветом. Качественная реакция на резорцин — появление темно-фиолетового окрашивания в присутствии хлорида трехвалентного железа. Постепенно цвет раствора становится черным. Формула реактива, который служит для распознавания фенола и некоторых его гомологов, — FeCl3 (хлорид железа (III)).
Гидроксибензол, нафтол, тимол — это все фенолы. Общая формула и состав веществ позволяет определить принадлежность этих соединение к ароматическому ряду. Все органические вещества, содержащие в своей формуле фенильный радикал С6Н5, с которым непосредственно связаны оксигруппы, проявляют особые свойства. От спиртов они отличаются лучше выраженным кислотным характером. По сравнению с веществами гомологического ряда бензола, фенолы — более активные химические соединения.
Урок по теме «Фенолы»
Образовательные задачи:
Главная: изучить состав, строение, свойства фенола и его соединений
Сопутствующие:
- на примере фенола конкретизировать знания учащихся об особенностях строения веществ, принадлежащих к классу фенолы, рассмотреть зависимость взаимного влияния атомов в молекуле фенола на его свойства
- познакомить учащихся с физическими и химическими свойствами фенола и некоторых его соединений, изучить качественные реакции на фенолы
- рассмотреть нахождение в природе, применение фенола и его соединений, их биологическую роль
Развивающие задачи:
- совершенствовать умение учащихся прогнозировать свойства вещества на основе его строения
- продолжать развивать умение наблюдать, анализировать, делать выводы при выполнении химического эксперимента
Воспитательные задачи:
- продолжить формирование химической картины мира через химическую картину природы (познаваемость, управление химическими процессами)
- расширить представление учащихся о влиянии фенолсодержащих промышленных отходов и строительных материалов на окружающую среду и здоровье человека
- рассмотреть биологическую роль фенола и его соединений на организм человека (положительную и отрицательную)
Тип урока:
урок — изучения новых знаний.
Методы обучения:
словесный, наглядный, практический (химический эксперимент – ученический и демонстрационный)
Средства обучения:
ТСО (графопроектор и кодосхемы), школьный химический эксперимент (демонстрационный и ученический), опорные конспекты, тестовые задания по материалам ЕГЭ (уровень А для работы на уроке, уровни В и С для выполнения дома)
Оборудование и реактивы:
Демонстрационный эксперимент: растворы С6Н5ОН, NaOH, FeCl3, бромная вода, HNO3(конц), Na, пробирки, резиновые пробки
Лабораторный опыт на определение наличия фенольных соединений в чае: пробирки с раствором чая, раствор FeCl3
План урока
1.Организационный момент
2.Актуализация знаний
3.Изучение новых знаний
? Какие спирты называются ароматическими
Работа по кодосхеме 1
? Будут ли отличаться химические свойства у
соединений, в одном из которых группа -ОН
связана непосредственно с бензольным ядром, а в
другом через атом углерода !!! Верно, согласно
одного из положений теории Бутлерова, которое
гласит, что свойства органических соединений
зависят не только от состава вещества и порядка
соединения атомов в молекуле, но и от взаимного
влияния атомов и групп атомов друг на друга.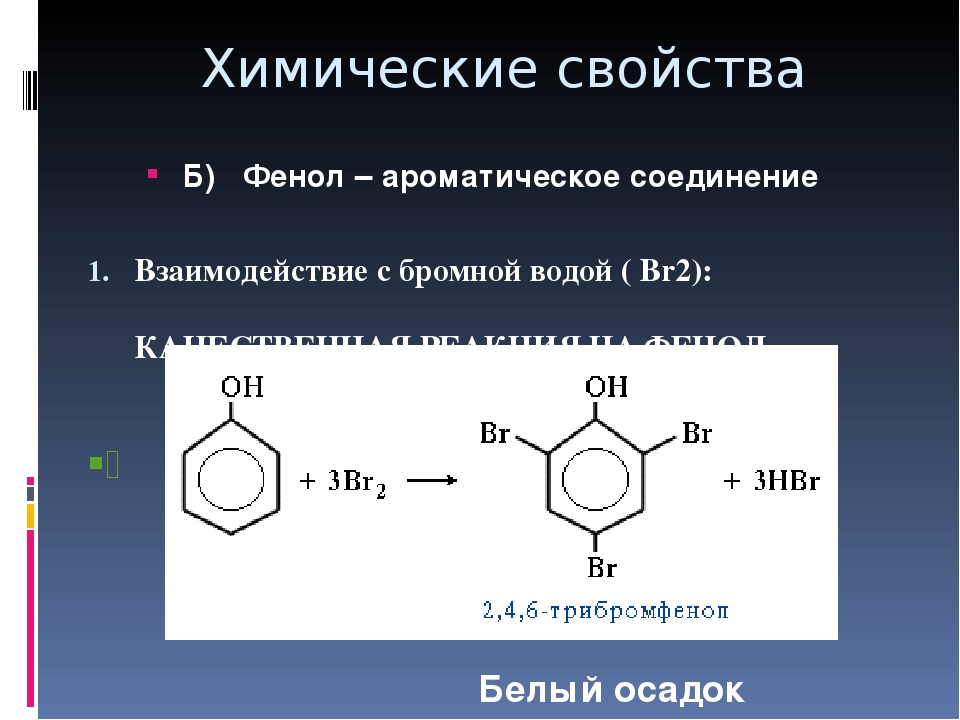 Причем, наибольшее влияние оказывают атомы
непосредственно связанные друг с другом. Влияние
атомов или групп атомов, не связанных
непосредственно ослабевает по мере их удаления
друг от друга.
Причем, наибольшее влияние оказывают атомы
непосредственно связанные друг с другом. Влияние
атомов или групп атомов, не связанных
непосредственно ослабевает по мере их удаления
друг от друга.
- Определение фенолов Соединения, в которых ароматический радикал фенил С6Н5- непосредственно связан с гидроксильной группой, отличаются по свойствам от ароматических спиртов, настолько, что их выделяют в отдельный класс органических соединений, называемый фенолами.
- классификация и изомерия фенолов В зависимости от числа ОН-групп различают одноатомные фенолы (например, вышеприведенные фенол и крезолы) и многоатомные. Среди многоатомных фенолов наиболее распространены двухатомные:
Как видно из приведенных примеров, фенолам
свойственна структурная изомерия (изомерия
положения гидроксигруппы).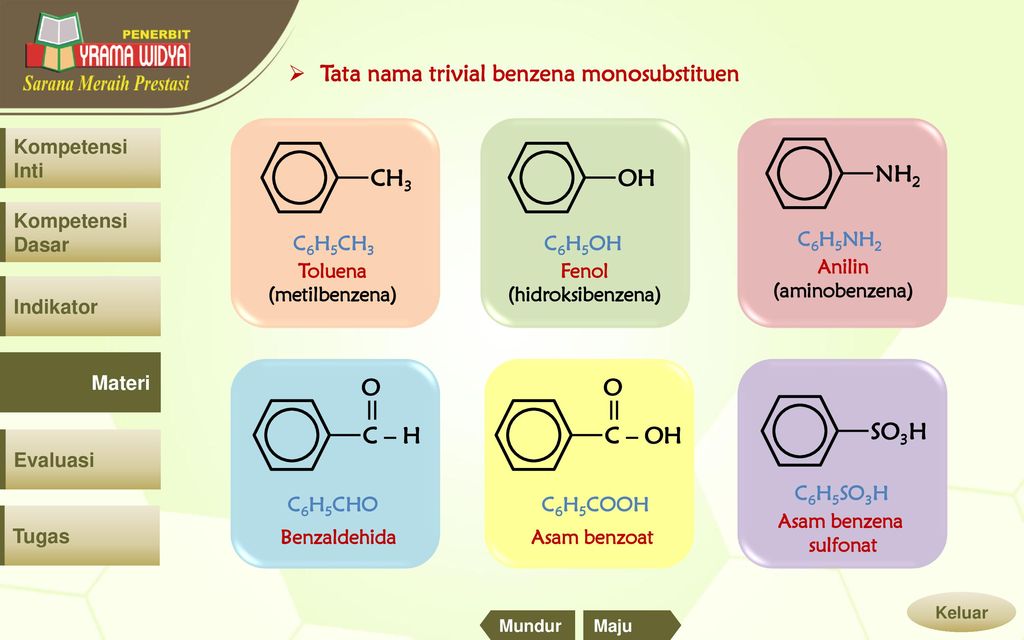
Следствием
полярности связи О–Н и наличия неподеленных пар
электронов на атоме кислорода является
способность гидроксисоединений к образованию водородных
связей
Это объясняет, почему у фенола довольно высокие температуры плавления (+43) и кипения (+182). Образование водородных связей с молекулами воды способствует растворимости гидроксисоединений в воде:
Способность растворяться в воде уменьшается с увеличением углеводородного радикала и от многоатомных гидроксисоединений к одноатомным. Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. Растворимость фенола в воде ограничена.
- Строение молекулы фенола
Кодосхема 2 |
|
 Фенол- более сильная
кислота, чем вода и спирты.
Фенол- более сильная
кислота, чем вода и спирты.- Химические свойства фенола (проводится демонстрационный эксперимент)
а) Рассмотрим реакции фенола по ОН-группе:
Кислотные свойства у фенола выражены сильнее, чем у спирта С2Н5ОН. Фенол – слабая кислота (карболовая).
б) Реакции фенола по бензольному кольцу:
Какой вывод о взаимном влиянии атомов в
молекуле фенола можно сделать?
Фенильная группа C6H5 – и гидроксил
–ОН взаимно влияют друг на друга. (кодосхема 3 )
(кодосхема 3 )
в) Качественная реакция на фенолы
С6Н5ОН + FeCl3 —> фиолетовое
окрашивание С6Н5ОН + Br2 —>
белый осадок С6Н4(ОН)2 + FeCl3
—> зеленое окрашивание,
С6Н3(ОН)3 + FeCl3 —> красное
окрашивание,
Проводится ученический эксперимент по определению наличия фенолов в чае
- Получение фенола самостоятельно (по опорному конспекту)
- Физиологическое действие фенола и его применение
Фенол — ядовит!!! При попадании на кожу вызывает ожоги, при этом он всасывается через кожу и вызывает отравление.
Биологическая роль соединений фенола (кодосхема)
Положительная |
Отрицательная (токсическое действие) |
|
|
- Первичное закрепление знаний ПОДУМАЙ и ОТВЕТЬ (задания части А ЕГЭ)
- Рефлексия
Домашнее задание § 18
что представляет вещество, его влияние на организм человека
Кислотно-основные свойства.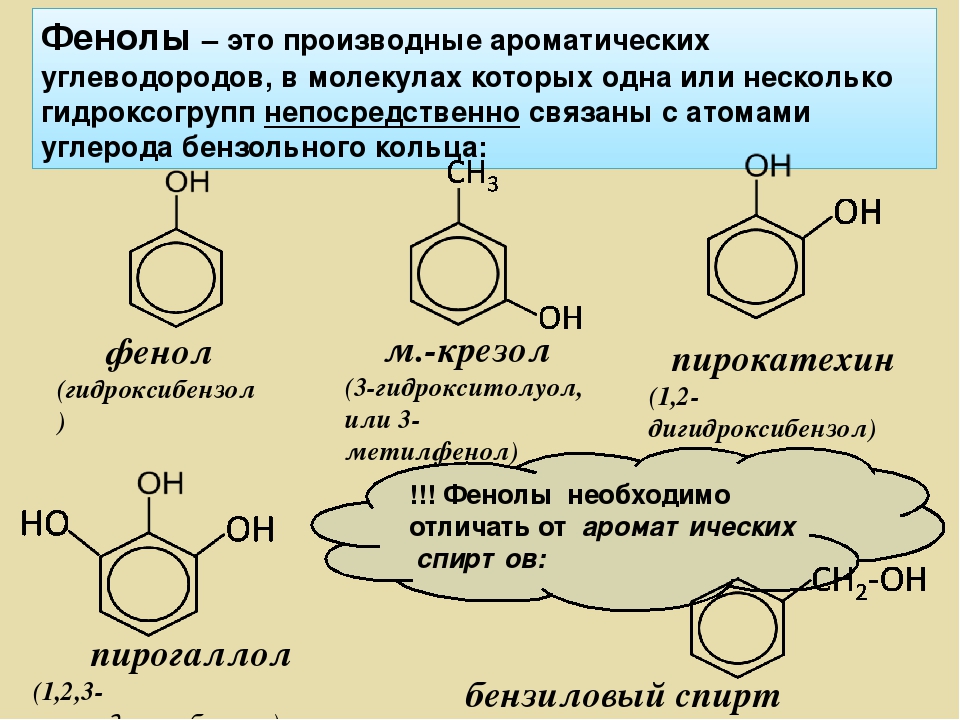 Кислотность фенолов значительно выше (на 5-6 порядков), чем кислотность спиртов. Это определяется двумя факторами: большей полярностью связи О-Н из-за того, что неподеленная электронная пара атома кислорода вовлечена в сопряжение с бензольным кольцом (гидроксильная группа — сильный донор по +М-эффекту), и значительной стабилизацией образующегося фенолят-иона за счет делокализации отрицательного заряда с участием ароматической системы:
Кислотность фенолов значительно выше (на 5-6 порядков), чем кислотность спиртов. Это определяется двумя факторами: большей полярностью связи О-Н из-за того, что неподеленная электронная пара атома кислорода вовлечена в сопряжение с бензольным кольцом (гидроксильная группа — сильный донор по +М-эффекту), и значительной стабилизацией образующегося фенолят-иона за счет делокализации отрицательного заряда с участием ароматической системы:
В отличие от алканолов фенолы при действии щелочей образуют соли — феноляты, растворимые в водных растворах щелочей (рН > 12). Однако фенолы плохо растворимы в водных растворах гидрокарбонатов щелочных металлов (рН = 8), так как в этих условиях феноляты подвергаются полному гидролизу.
Основные свойства фенола выражены значительно слабее (на 4-5 порядков), чем у спиртов. Это связано с тем, что сопряжение неподеленной электронной пары кислородного атома с π-электро-нами бензольного кольца в образующемся катионе нарушено:
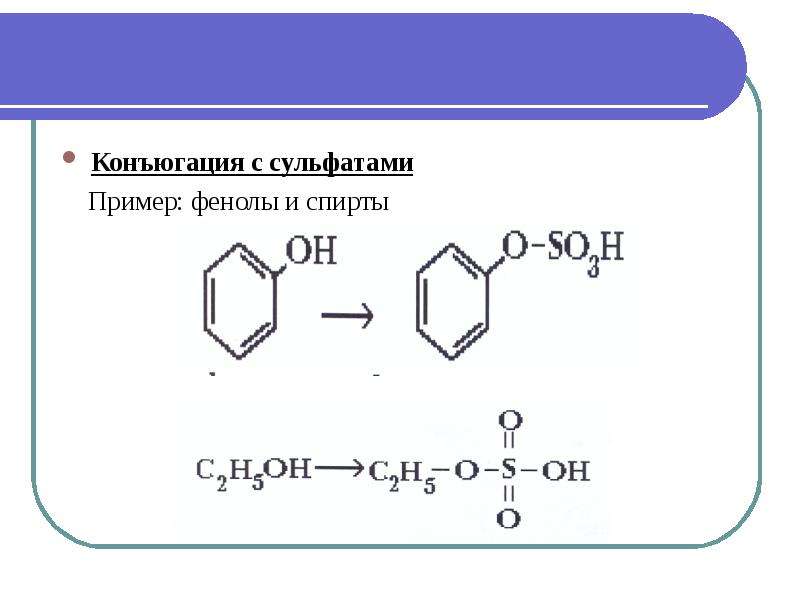
Ацилирование. Этерификация карбоновыми кислотами в присутствии h3SO4, характерная для спиртов, в случае фенола идет медленно из-за низкой нуклеофильности его кислородного центра. Поэтому для получения сложных эфиров фенола применяют более сильные электрофилы — хлорангидриды RC0C1 или ангидриды [(RCO) 2 0] карбоновых кислот в безводных условиях:
Этерификация карбоновыми кислотами в присутствии h3SO4, характерная для спиртов, в случае фенола идет медленно из-за низкой нуклеофильности его кислородного центра. Поэтому для получения сложных эфиров фенола применяют более сильные электрофилы — хлорангидриды RC0C1 или ангидриды [(RCO) 2 0] карбоновых кислот в безводных условиях:
Алкилирование фенола. Нуклеофильность кислородного центра в фенолятах значительно выше, чем в феноле. Так, при обработке фенолята натрия галоидными алкилами образуются простые эфиры фенолов:
Все рассмотренные реакции фенолов происходят по связи О-Н. Реакции с разрывом связи С-О в фенолах, т. е. реакции замещения гидроксильной группы в феноле, в организме не происходят.
Окислительно-восстановительные свойства. Фенол легко окисляется на воздухе, из-за чего его белые кристаллы быстро розовеют. Состав образующихся продуктов точно не установлен.
Фенолы имеют характерную цветную реакцию с FeCl3 в водных растворах с появлением красно-фиолетового окрашивания, которое исчезает после прибавления сильной кислоты или спирта. Предполагают, что интенсивная окраска связана с образованием комплексного соединения, содержащего во внутренней сфере фенолят-анион:
Предполагают, что интенсивная окраска связана с образованием комплексного соединения, содержащего во внутренней сфере фенолят-анион:
В этом комплексе из всех лигандов фенолят-анион — самый активный нуклеофил и восстановитель. Он способен передать один электрон электрофилу и окислителю — катиону железа(3) — с образованием во внутренней сфере ион-радикальной системы, содержащей феноксильный радикал (C6H5O*), что приводит к появлению интенсивной окраски:
Подобное образование радикалов во внутренней сфере комплексного соединения за счет внутрисферного окислительно-восстановительного процесса может происходить и в субстрат-ферментных комплексах организма. При этом радикальная частица может или оставаться связанной во внутренней сфере, или становиться свободной при выходе из этой сферы.
Рассмотренная реакция с FeCl3 свидетельствует о легкости окисления фенола, особенно его аниона. Еще легче окисляются многоатомные фенолы. Так, гидрохинон (особенно его дианион) легко окисляется за счет углеродных атомов в 1,4-бензохинон:
Гидрохинон используется в фотографии, поскольку он. восстанавливает AgBr в фотографической эмульсии на засвеченных участках быстрее, чем на незасвеченных.
восстанавливает AgBr в фотографической эмульсии на засвеченных участках быстрее, чем на незасвеченных.
Соединения, содержащие 1,4-хиноидную группировку, называют хинонами. Хиноны — типичные окислители, образующие с соответствующими гидрохинонами равновесную сопряженную окислительно-восстановительную пару (разд. 9.1). Такая пара в коферменте Q участвует в процессе окисления субстрата за счет дегидрирования (разд. 9.3.3) и переноса электронов по электронотранспортной цепи от окисляемого субстрата к кислороду (разд. 9.3.4). Витамины группы К, содержащие нафтохиноновую группировку, обеспечивают свертывание крови на воздухе.
Электрофильное замещение по бензольному кольцу. Благодаря электронодонорному эффекту гидроксильной группы фенол значительно легче вступает в реакции электрофильного замещения, чем бензол. Гидроксильная группа ориентирует атаку электрофила в о- и n-положения. Например, фенол обесцвечивает бромную воду при комнатной температуре с образованием 2,4,6-трибромфенола:
Активность фенола в реакциях электрофильного замещения настолько велика, что он реагирует даже с альдегидами.
 Эта реакция поликонденсации лежит в основе получения различных фенолоформальдегидных смол, широко используемых в промышленности. При проведении поликонденсации в кислой среде образуются бакелитовые полимеры,
а в щелочной среде, где реакция идет глубже из-за высокой активности фенолят-аниона, — резольные полимеры:
Эта реакция поликонденсации лежит в основе получения различных фенолоформальдегидных смол, широко используемых в промышленности. При проведении поликонденсации в кислой среде образуются бакелитовые полимеры,
а в щелочной среде, где реакция идет глубже из-за высокой активности фенолят-аниона, — резольные полимеры:
Важнейшие представители спиртов и их практическое значение. Алканолы — физиологически активные вещества, обладающие наркотическим действием. Это действие возрастает с разветвлением и удлинением углеродной цепи, проходя через максимум при C6-C8, а также при переходе от первичных спиртов к вторичным. Продукты превращения спиртов в организме могут служить причиной их токсического действия.
Метанол СН 3 ОН — сильный яд, так как в пищеварительном тракте окисляется в формальдегид и муравьиную кислоту. Уже в небольших дозах (10 мл) может вызвать слепоту.
Этанол С2Н5ОН, обычно называемый просто спирт. Употребление этанола (алкогольных напитков) действует вначале возбуждающе, а затем угнетающе на центральную нервную систему, притупляет чувствительность, ослабляет функцию мозга и мышечной системы, ухудшает реакцию.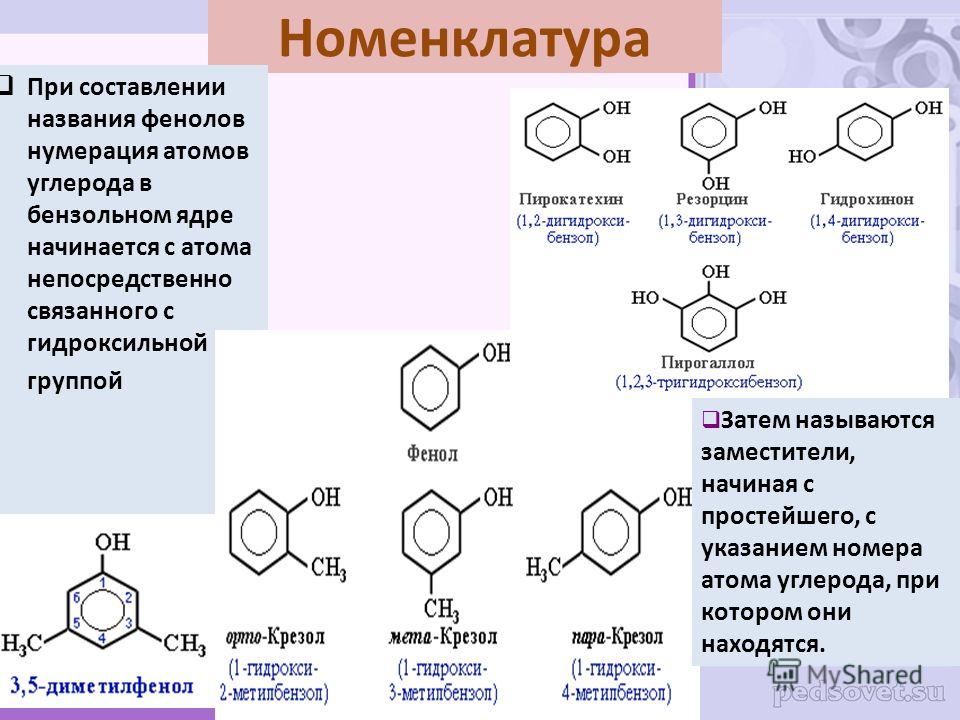 Его длительное и неумеренное употребление приводит к алкоголизму. Механизм действия этанола на организм чрезвычайно сложен и окончательно еще не выяснен. Однако важной стадией его превращения в организме является образование ацетальдегида, который легко реагирует со многими важными метаболитами.
Его длительное и неумеренное употребление приводит к алкоголизму. Механизм действия этанола на организм чрезвычайно сложен и окончательно еще не выяснен. Однако важной стадией его превращения в организме является образование ацетальдегида, который легко реагирует со многими важными метаболитами.
Этиленгликоль НОСН2СН2ОН — сильный яд, так как продуктами его превращения в организме являются щавелевая кислота и другие не менее ядовитые соединения. Обладает спиртовым запахом, в связи с чем может быть принят за этанол и явиться причиной тяжелых интоксикаций. Используется в технике как антиобледенитель и для приготовления антифризов -жидкостей с низкой температурой замерзания, применяемых для охлаждения двигателей зимой.
Глицерин НОСН 2 СН(ОН)СН 2 ОН — нетоксичная, вязкая, бесцветная жидкость сладкого вкуса. Он входит в состав большинства омыляемых липидов: животных и растительных жиров, а также фосфолипидов. Применяется для производства тринитрата глицерина, в качестве мягчителя в текстильной и кожевенной промышленности и как составная часть косметических препаратов для смягчения кожи.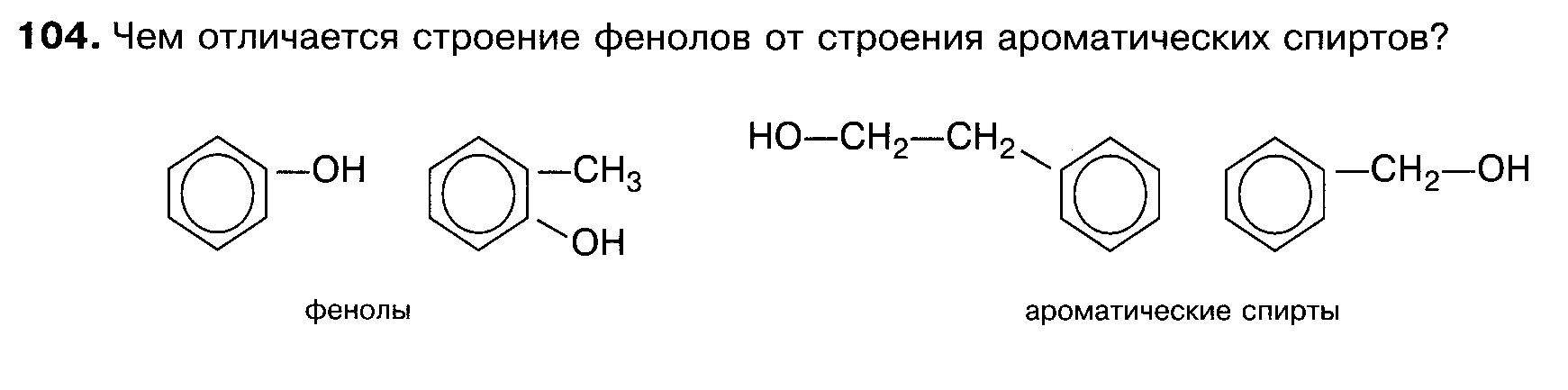
Биологически активными спиртами являются многие метаболиты, относящиеся к разным классам органических соединений: ментол — класс терпенов; ксилит, сорбит, мезоинозит -многоатомные спирты; холестерин, эстрадиол — стероиды.
Можно ожидать, что в зависимости от этого свойства веществ будут существенно отличаться друг от друга из-за взаимного влияния групп атомов (вспомните одно из положений теории Бутлерова). И действительно, органические соединения, содержащие ароматический радикал фенил С 6 Н 5 -, непосредственно связанный с гидроксильной группой, проявляют особые свойства, отличные от свойств спиртов. Такие соединения называют фенолами.
— органические вещества, молекулы которых содержат радикал фенил, связанный с одной или несколькими гидроксигруппами.
Так же как и спирты, фенолы классифицируют по атомности, т. е. по количеству гидроксильных групп.
Одноатомные фенолы содержат в молекуле одну гидроксильную группу:
Существуют и другие многоатомные фенолы
, содержащие три и более гидроксиль-ные группы в бензольном кольце.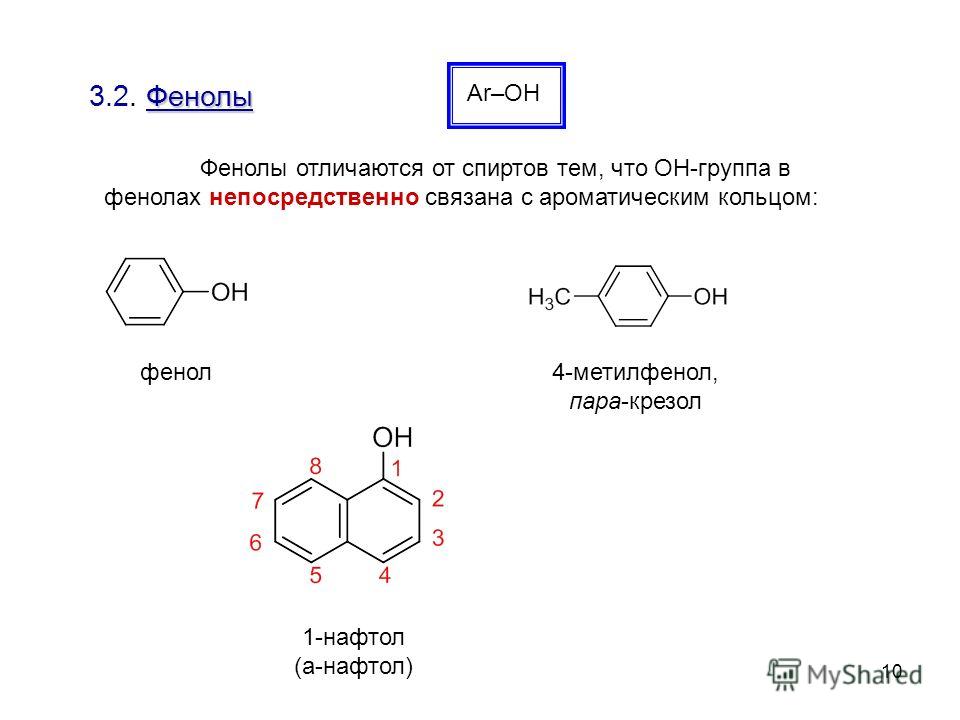
Познакомимся подробнее со строением и свойствами простейшего представителя этого класса — фенолом С6Н50Н. Название этого вещества и легло в основу названия всего класса — фенолы.
Физические свойства
Твердое бесцветное кристаллическое вещество, tºпл = 43 °С, tº кип = °С, с резким характерным запахом. Ядовит. Фенол при комнатной температуре незначительно растворяется в воде. Водный раствор фенола называют карболовой кислотой. При попадании на кожу он вызывает ожоги, поэтому с фенолом необходимо обращаться осторожно.
Строение молекулы фенола
В молекуле фенола гидроксил непосредственно связан с атомом углерода бензольного ароматического ядра.
Вспомним строение групп атомов, образующих молекулу фенола.
Ароматическое кольцо состоит из шести атомов углерода, образующих правильный шестиугольник, вследствие,sр 2 -гибридизации электронных орбиталей шести атомов углерода. Эти атомы связаны Þ-связями. Не участвующие в образовании ст-связей р-электроны каждого атома углерода, перекрывающиеся по разные стороны плоскости Þ-связей, образуют две части единого шестиэлектронного п
-облака, охватывающего все бензольное кольцо (ароматическое ядро). В молекуле бензола С6Н6 ароматическое ядро абсолютно симметрично, единое электронное п
-облако равномерно охватывает кольцо атомов углерода под и над плоскостью молекулы (рис. 24).
В молекуле бензола С6Н6 ароматическое ядро абсолютно симметрично, единое электронное п
-облако равномерно охватывает кольцо атомов углерода под и над плоскостью молекулы (рис. 24).
Ковалентная связь между атомами кислорода и водорода гидроксиль-ного радикала сильно полярна, общее электронное облако связи О-Н смещено в сторону атома кислорода , на котором возникает частичный отрицательный заряд, а на атоме водорода — частичный положительный заряд. Кроме того, атом кислорода в гидроксильной группе имеет две неподеленные, принадлежащие только ему электронные пары.
В молекуле фенола гидроксильный радикал взаимодействует с ароматическим ядром, при этом неподеленные электронные пары атома кислорода взаимодействуют с единым тс-облаком бензольного кольца, образуя единую электронную систему. Такое взаимодействие неподеленных электронных пар и облаков тг-связей называют сопряжением. В результате сопряжения неподеленной электронной пары атома кислорода гидроксигруппы с электронной системой бензольного кольца уменьшается электронная плотность на атоме кислорода. Это снижение компенсируется за счет большей поляризации связи О-Н, что, в свою очередь, приводит к увеличению положительного заряда на атоме водорода. Следовательно, водород гидроксильной группы в молекуле фенола имеет «кислотный» характер.
Это снижение компенсируется за счет большей поляризации связи О-Н, что, в свою очередь, приводит к увеличению положительного заряда на атоме водорода. Следовательно, водород гидроксильной группы в молекуле фенола имеет «кислотный» характер.
Логично предположить, что сопряжение электронов бензольного кольца и гидроксильной группы сказывается не только на ее свойствах, но и на реакционной способности бензольного кольца.
В самом деле, как вы помните, сопряжение неподеленных пар атома кислорода с л-облаком бензольного кольца приводит к перераспределению электронной плотности в нем. Она понижается у атома углерода, связанного с ОН-группой (сказывается влияние электронных пар атома кислорода) и повышается у соседних с ним атомов углерода (т. е. положения 2 и 6, или орто-положения). Очевидно, что повышение электронной плотности у этих атомов углерода бензольного кольца приводит к локализации (сосредоточению) отрицательного заряда на них. Под влиянием этого заряда происходит дальнейшее перераспределение электронной плотности в ароматическом ядре — смещение ее от 3-го и 5-го атомов (.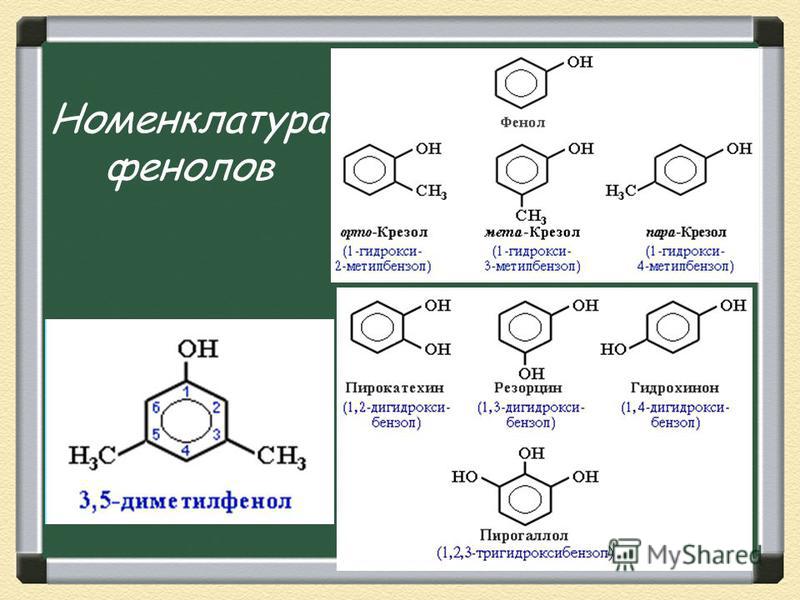 мета-положение) к 4-му (орто-положение). Эти процессы можно выразить схемой:
мета-положение) к 4-му (орто-положение). Эти процессы можно выразить схемой:
Таким образом, наличие гидроксильного радикала в молекуле фенола приводит к изменению л-облака бензольного кольца, увеличению электронной плотности у 2, 4 и 6-го атомов углерода (орто-, дара-положения) и уменьшению электронной плотности у 3-го и 5-го атомов углерода (мета-положения).
Локализация электронной плотности в орто- и пара-положениях делает их наиболее вероятными для атак электрофильных частиц при взаимодействии с другими веществами.
Следовательно, влияние радикалов, составляющих молекулу фенола, взаимно, и оно определяет его характерные свойства.
Химические свойства фенола
Кислотные свойства
Как уже было сказано, атом водорода гидроксильной группы фенола обладает кислотным характером. Кислотные свойства у фенола выражены сильнее, чем у воды и спиртов . В отличие от спиртов и воды фенол реагирует не только с щелочными металлами, но и с щелочами с образованием фенолятов.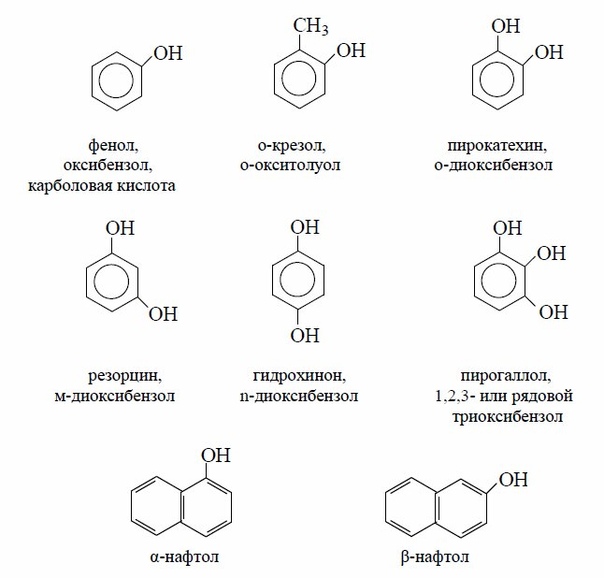
Однако кислотные свойства у фенолов выражены слабее, чем у неорганических и карбоновых кислот. Так, например, кислотные свойства фенола примерно в 3000 раз меньше, чем у угольной кислоты. Поэтому, пропуская через водный раствор фенолята натрия углекислый газ, можно выделить свободный фенол:
Добавление к водному раствору фенолята натрия соляной или серной кислоты также приводит к образованию фенола.
Качественная реакция на фенол
Фенол реагирует с хлоридом железа(ІІІ) с образованием интенсивно окрашенного в фиолетовый цвет комплексного соединения.
Эта реакция позволяет обнаруживать его даже в очень незначительных количествах. Другие фенолы, содержащие одну или несколько гидроксильных групп в бензольном кольце, также дают яркое окрашивание сине-фиолетовых оттенков в реакции с хлоридом железа(ІІІ).
Реакции бензольного кольца
Наличие гидроксильного заместителя значительно облегчает протекание реакций электрофильного замещения в бензольном кольце.
1. Бромирование фенола. В отличие от бензола для бромирования фенола не требуется добавления катализатора (бромида железа(ІІІ)).
Кроме того, взаимодействие с фенолом протекает селективно (избирательно): атомы брома направляются в орто- и пара-положения, замещая находящиеся там атомы водорода. Селективность замещения объясняется рассмотренными выше особенностями электронного строения молекулы фенола. Так, при взаимодействии фенола с бромной водой образуется белый осадок 2,4,6-трибромфенола.
Эта реакция, так же как и реакция с хлоридом железа(ІІІ), служит для качественного обнаружения фенола.
2. Нитрование фенола также происходит легче, чем нитрование бензола. Реакция с разбавленной азотной кислотой идет при комнатной температуре. В результате образуется смесь орто- и пара-изомеров нитрофенола:
3. Гидрирование ароматического ядра фенола в присутствии катализатора происходит легко.
4. Поликонденсация фенола с альдегидами, в частности, с формальдегидом, происходит с образованием продуктов реакции — фенолформальдегидных смол и твердых полимеров.
Взаимодействие фенола с формальдегидом можно описать схемой:
Вы, наверное, заметили, что в молекуле димера сохраняются «подвижные» атомы водорода, а значит, возможно дальнейшее продолжение реакции при достаточном количестве реагентов.
Реакция поликонденсации, т. е. реакция получения полимера, протекающая с выделением побочного низкомолекулярного продукта (воды), может продолжаться и далее (до полного израсходования одного из реагентов) с образованием огромных макромолекул. Процесс можно описать суммарным уравнением:
Образование линейных молекул происходит при обычной температуре. Проведение же этой реакции при нагревании приводит к тому, что образующийся продукт имеет разветвленное строение, он твердый и нерастворимый в воде. В результате нагревания феноло-формальдегидной смолы линейного строения с избытком альдегида получаются твердые пластические массы с уникальными свойствами. Полимеры на основе феноло-формальдегидных смол применяют для изготовления лаков и красок, пластмассовых изделий, устойчивых к нагреванию, охлаждению, действию воды, щелочей и кислот, они обладают высокими диэлектрическими свойствами. Из полимеров на основе фенолформальдегидных смол изготавливают наиболее ответственные и важные детали электроприборов, корпуса силовых агрегатов и детали машин, полимерную основу печатных плат для радиоприборов.
Из полимеров на основе фенолформальдегидных смол изготавливают наиболее ответственные и важные детали электроприборов, корпуса силовых агрегатов и детали машин, полимерную основу печатных плат для радиоприборов.
Клеи на основе феноло-формальдегидных смол способны надежно соединять детали самой различной природы, сохраняя высочайшую прочность соединения в очень широком диапазоне температур. Такой клей применяется для крепления металлического цоколя ламп освещения к стеклянной колбе. Теперь вам стало понятно, почему фенол и продукты на его основе находят широкое применение (схема 8).
1. Назовите вещества по их структурным формулам:
2. Объясните, почему кислотные свойства фенола выражены сильнее, чем кислотные свойства воды и спиртов.
3. При пропускании углекислого газа через водный раствор фенолята натрия реакционная смесь помутнела и приобрела характерный запах. Объясните изменения и приведите уравнения реакций в молекулярном, полном и сокращенном ионном виде.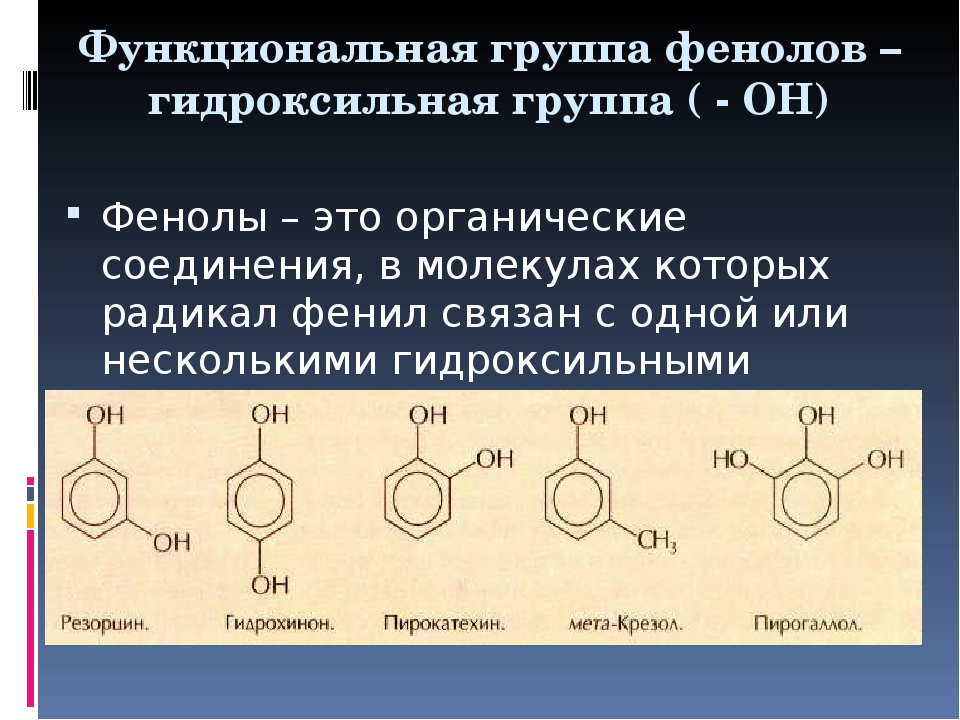
4. Составьте уравнения реакций, соответствующих нескольким стадиям образования фенолформальдегидного полимера из тримера.
5*. Смесь непредельного спирта и гомолога фенола массой 1,37 г реагирует с 160 г 2%-ной бромной воды. Такая же смесь в реакции с избытком натрия выделяет 168 мл газа (н. у.). Определите молекулярные формулы веществ и их массовые доли в смеси.
Это вещество было открыто в 1771
году. Сразу после открытия его стали использовать в качестве красителя. Текстильщики красили им свои ткани. В 1834
году немецкий химик Фридлиб Рунге
обнаружил в продуктах перегонки каменноугольной смолы белое кристаллическое вещество с характерным запахом, но ему не удалось определить его состав. И только в 1841
году Огюст Лоран
установил его формулу.
Сразу после открытия его стали использовать в качестве красителя. Текстильщики красили им свои ткани. В 1834
году немецкий химик Фридлиб Рунге
обнаружил в продуктах перегонки каменноугольной смолы белое кристаллическое вещество с характерным запахом, но ему не удалось определить его состав. И только в 1841
году Огюст Лоран
установил его формулу.
- Определение фенолов.
- Классификация и изомерия фенолов.
Как видно из приведенных примеров, фенолам свойственна структурная изомерия (изомерия положения гидроксигруппы).
- Физические свойства фенола.
Это объясняет, почему у фенола довольно высокие температуры плавления (+43 ) и кипения (+182 ). Образование водородных связей с молекулами воды способствует растворимости гидроксисоединений в воде:
Способность растворяться в воде уменьшается с увеличением углеводородного радикала и от многоатомных гидроксисоединений к одноатомным. Метанол, этанол, пропанол, изопропанол, этиленгликоль и глицерин смешиваются с водой в любых соотношениях. Растворимость фенола в воде ограничена.
Для более полного представления о физических свойствах посмотрите видеоролик:
- Строение молекулы фенола.
- неподеленная электронная пара атома кислорода притягивается 6-ти электронным облаком бензольного кольца, из – за чего связь О – Н еще сильнее поляризуется.
 Фенол- более сильная кислота, чем вода и спирты.
Фенол- более сильная кислота, чем вода и спирты.
- В бензольном кольце нарушается симметричность электронного облака, электронная плотность повышается в положении 2, 4, 6. Это делает более реакционноспособными связи С — Н в положениях 2, 4, 6. и? – связи бензольного кольца.
- Химические свойства фенола.
а) кислотные свойства:
Кислотность фенола существенно выше, чем у предельных спиртов; он реагирует как с щелочными металлами,
так и с их гидроксидами (отсюда старинное название «карболовая кислота»):
Кислотные свойства у фенола выражены сильнее, чем у спирта С 2 Н 5 ОН. Фенол – слабая кислота (карболовая).
Фенол, однако, является очень слабой кислотой. При пропускании через раствор фенолятов углекислого или сернистого газов выделяется фенол; такая реакция доказывает, что фенол — более слабая кислота, чем угольная и сернистая:
C
6
H
5
ONa
+ СО 2 + Н 2 О →
С 6 Н 5 ОН +
NaHCO
3
.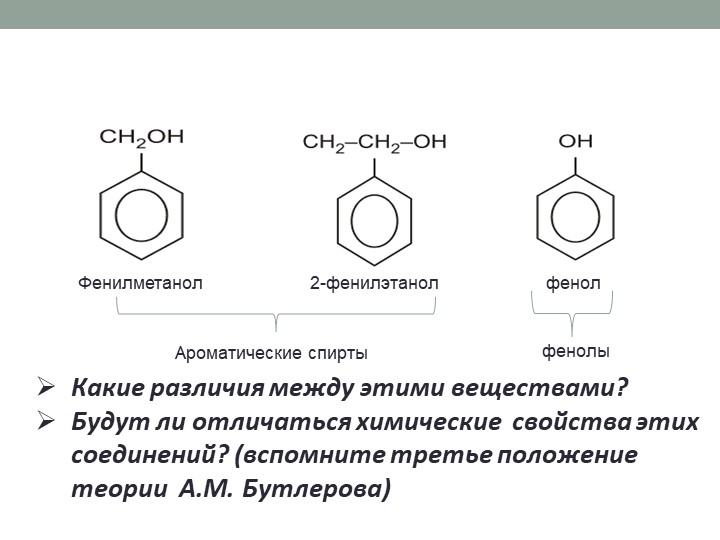
б) Образование сложных эфиров.
В отличие от спиртов, фенолы не образуют сложных эфиров при действии на них карбоновых кислот; для этого используются хлорангидриды кислот:
С 6 Н 5 ОН + СН 3 ― CO ― Cl → С 6 Н 5 ― О― СО― СН 3 + HCl .
II. Реакции фенола по бензольному кольцу:
- взаимодействие с бромной водой:
- взаимодействие с азотной кислотой:
При нитровании фенола концентрированной азотной кислотой три атома водорода замещаются на нитрогруппу, и образуется 2,4,6-тринитрофенол (пикриновая кислота):
- реакция поликонденсации
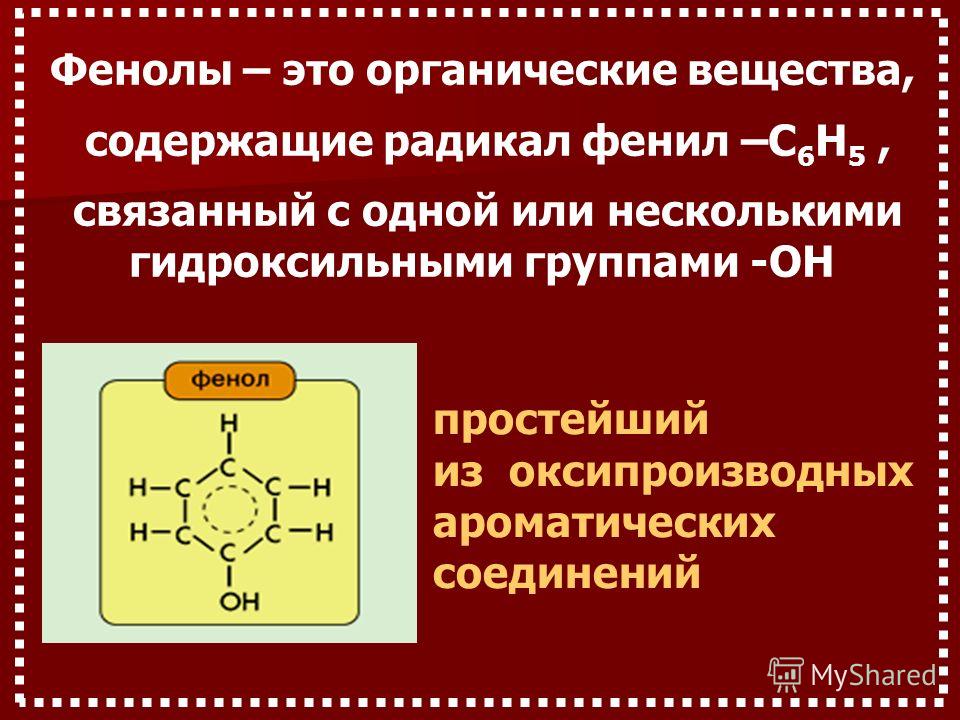 Качественная реакция на фенолы
Качественная реакция на фенолы
- С 6 Н 5 ОН + FeCl 3 —> фиолетовое окрашивание
- С 6 Н 5 ОН + Br 2 —> белый осадок
- С 6 Н 4 (ОН) 2 + FeCl 3 —>зеленое окрашивание
- С 6 Н 3 (ОН) 3 + FeCl 3 —> красное окрашивание
IV. Окисление.
Фенолы легко окисляются
даже под действием кислорода воздуха. Так, при стоянии на воздухе фенол
постепенно окрашивается в розовато-красный цвет. При энергичном
окислении фенола хромовой смесью основным продуктом окисления является
хинон. Двухатомные фенолы окисляются еще легче. При окислении
гидрохинона
также образуется хинон
:
- Получение фенола.
1 . Получение из галогенбензолов . При нагревании хлорбензола и гидроксида натрия под давлением получают фенолят натрия, при дальнейшей обработке которого кислотой образуется фенол:
С 6 Н 5 ―
С
l
+ 2
NaOH
→
C
6
H
5
―
ONa
+
NaCl
+ Н 2 О.
(1)
Это — основной промышленный способ получения фенола.3. Получение из ароматических сульфокислот. Реакция проводится при сплавлении сульфокислот с щелочами. Первоначально образующиеся феноксиды обрабатывают сильными кислотами для получения свободных фенолов. Метод обычно применяют для получения многоатомных фенолов:
Химические свойства фенолов определяются наличием в молекуле гидроксильной группы и бензольного кольца.
Реакции по гидроксильной группе
Фенолы, так же, как и алифатические спирты, обладают кислыми свойствами, т.е. способны образовывать соли – феноляты . Однако они более сильные кислоты и поэтому могут взаимодействовать не только со щелочными металлами (натрий, литий, калий), но и со щелочами и карбонатами:
Константа
кислотности рК
а
фенола равна 10.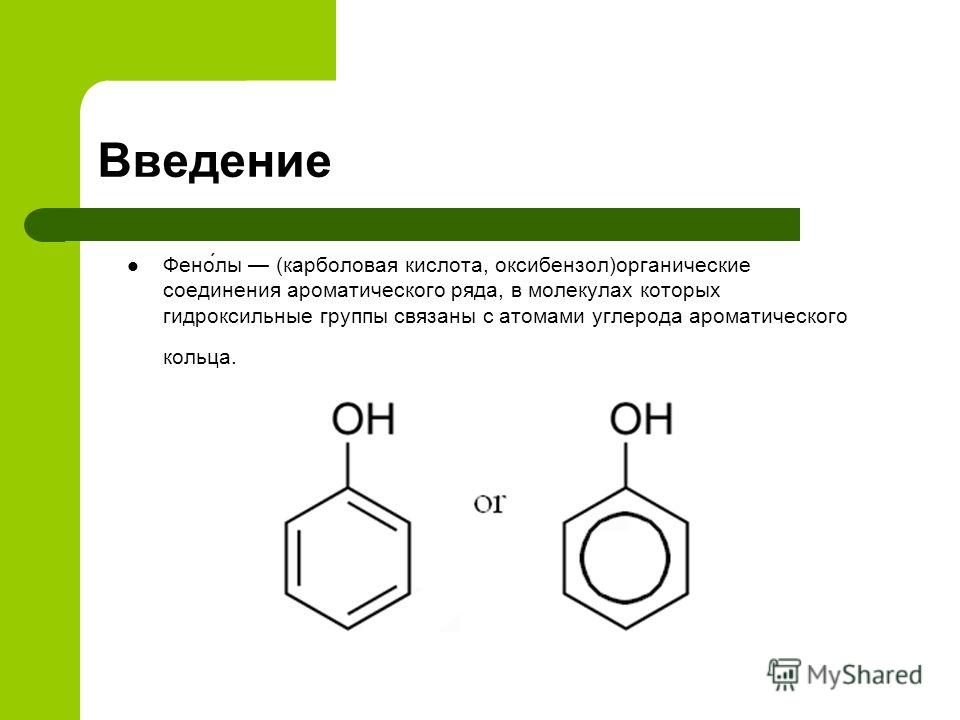 Высокая кислотность
фенола связана с акцепторным свойством
бензольного кольца (эффект
сопряжения
)
и объясняется резонансной стабилизацией
образующегося фенолят-аниона. Отрицательный
заряд на атоме кислорода фенолят-аниона
за счет эффекта сопряжения может
перераспределяться по ароматическому
кольцу, этот процесс можно описать
набором резонансных структур:
Высокая кислотность
фенола связана с акцепторным свойством
бензольного кольца (эффект
сопряжения
)
и объясняется резонансной стабилизацией
образующегося фенолят-аниона. Отрицательный
заряд на атоме кислорода фенолят-аниона
за счет эффекта сопряжения может
перераспределяться по ароматическому
кольцу, этот процесс можно описать
набором резонансных структур:
Ни одна из этих структур в отдельности не описывает реального состояния молекулы, но их использование позволяет объяснять многие реакции.
Феноляты легко взаимодействуют с галогеналканами и галогенангидридами:
Взаимодействие солей фенола с галогеналканами – реакция О-алкилирования фенолов. Это способ получения простых эфиров (реакция Вильямсона, 1852 г.).
Фенол способен взаимодействовать с галогенангидридами и ангидридами кислот с получением сложных эфиров (О-ацилирование):
Реакция протекает в присутствии небольших количеств минеральной кислоты или при нагревании.
Реакции по бензольному кольцу
Гидроксил является электронодонорной группой и активирует орто — и пара -положения в реакциях электрофильного замещения:
Галогенирование
Галогенирование фенолов действием галогенов или галогенирующих агентов протекает с большой скоростью:
Нитрование
При действии азотной кислоты в уксусной кислоте (в присутствии небольшого количества серной кислоты) на фенол получается 2-нитрофенол:
Под
действием концентрированной азотной
кислоты или нитрующей смеси фенол
интенсивно окисляется, что приводит к
глубокой деструкции его молекулы. При
использовании разбавленной азотной
кислоты нитрование сопровождается
сильным осмолением несмотря на охлаждение
до 0°С и приводит к образованию о-
и п-
изомеров
с преобладанием первого из них:
При
использовании разбавленной азотной
кислоты нитрование сопровождается
сильным осмолением несмотря на охлаждение
до 0°С и приводит к образованию о-
и п-
изомеров
с преобладанием первого из них:
При нитровании фенола тетраоксидом диазота в инертном растворителе (бензол, дихлорэтан) образуется 2,4-динитрофенол:
Нитрование последнего нитрующей смесью протекает легко и может служить методом синтеза пикриновой кислоты:
Эта реакция идет с саморазогреванием.
Пикриновую кислоту получают также через стадию сульфирования. Для этого обрабатывают фенол при 100°С избыточным количеством серной кислоты, получают 2,4-дисульфопроизводное, которое не выделяя из реакционной меси обрабатывают дымящей азотной кислотой:
Введение двух
сульфогрупп (также как и нитрогрупп) в
бензольное ядро делает его устойчивым
к окисляющему действию дымящей азотной
кислоты, реакция не сопровождается
осмолением. Такой метод получения
пикриновой кислоты удобен для производства
в промышленном масштабе.
Сульфирование . Сульфирование фенола в зависимости от температуры протекает в орто — или пара -положение:
Алкилирование и ацилирование по Фриделю-Крафтсу . Фенолы образуют с хлористым алюминием неактивные соли ArOAlCl 2 , поэтому для алкилирования фенолов в качестве катализаторов применяют протонные кислоты (H 2 SO 4) или металлооксидные катализаторы кислотного типа (Al 2 O 3). Это позволяет использовать в качестве алкилирующих агентов только спирты и алкены:
Алкилирование протекает последовательно с образованием моно-, ди- и триалкилфенолов. Одновременно происходит кислотно-катализируемая перегруппировка с миграцией алкильных групп:
Конденсация
с альдегидами и кетонами
.
При действии щелочных или кислотных
катализаторов на смесь фенола и альдегида
жирного ряда происходит конденсация в
о
—
и п
-положениях.
Эта реакция имеет очень большое
практическое значение, так как лежит в
основе получения важных пластических
масс и лаковых основ.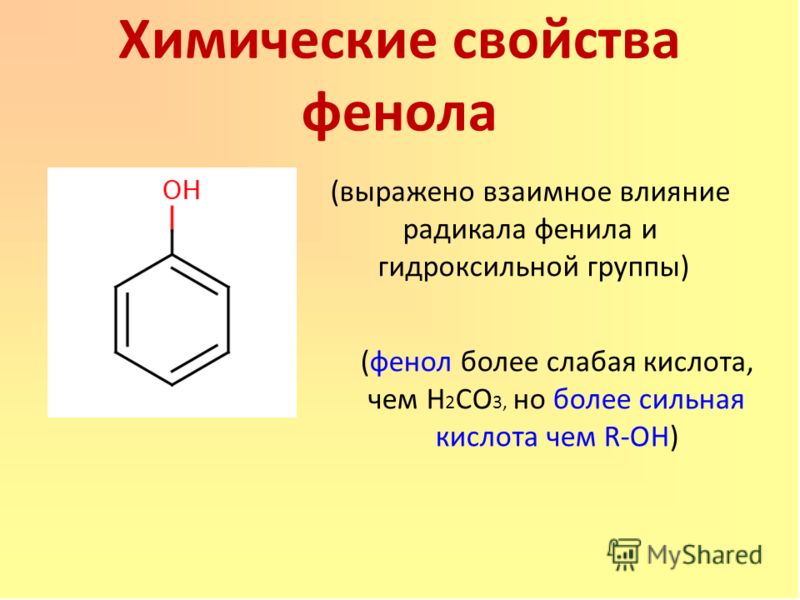 При обычной
температуре рост молекулы за счет
конденсации идет в линейном направлении:
При обычной
температуре рост молекулы за счет
конденсации идет в линейном направлении:
Если реакцию проводить при нагревании, начинается конденсация с образованием разветвленных молекул:
В результате присоединения по всем доступным о — и п -положениям образуется трехмерный термореактивный полимер – бакелит. Бакелит отличается высоким электрическим сопротивлением и термостойкостью. Это один из первых промышленных полимеров.
Реакция фенола с ацетоном в присутствии минеральной кислоты приводит к получению бисфенола:
Последний используют для получения эпоксисоединений.
Реакция Кольбе – Шмидта. Синтез фенилкарбоновых кислот.
Феноляты натрия и калия реагируют с углекислым газом, образуя в зависимости от температуры орто- или пара-изомеры фенилкарбоновых кислот:
Окисление
Фенол легко окисляется под действием хромовой кислоты до п -бензохинона:
Восстановление
Восстановление фенола в циклогексанон используют для получения полиамида (найлон-6,6)
Образованные на основе бензола. При нормальных условиях представляют собой твердые ядовитые вещества, обладающие специфическим ароматом. В современной промышленности эти химические соединения играют далеко не последнюю роль. По объемам использования фенол и его производные входят в двадцатку наиболее востребованных химических соединений в мире. Они широко применяются в химической и легкой промышленности, фармацевтике и энергетике. Поэтому получение фенола в промышленных масштабах — одна из основных задач химической промышленности.
При нормальных условиях представляют собой твердые ядовитые вещества, обладающие специфическим ароматом. В современной промышленности эти химические соединения играют далеко не последнюю роль. По объемам использования фенол и его производные входят в двадцатку наиболее востребованных химических соединений в мире. Они широко применяются в химической и легкой промышленности, фармацевтике и энергетике. Поэтому получение фенола в промышленных масштабах — одна из основных задач химической промышленности.
Обозначения фенола
Первоначальное название фенола — карболовая кислота. Позднее данное соединение поучило название «фенол». Формула этого вещества представлена на рисунке:
Нумерация атомов фенола ведется от того атома углерода, который соединен с гидроксогруппой ОН. Последовательность продолжается в таком порядке, чтобы другие замещенные атомы получили наименьшие номера. Производные фенола существуют в виде трех элементов, характеристики которых объясняются различием их структурных изомеров.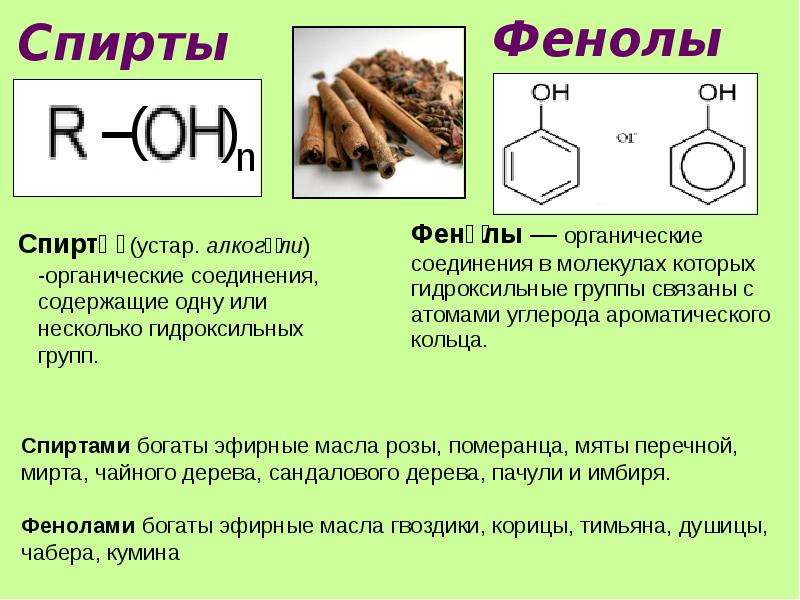 Различные орто-, мета-, паракрезолы являются лишь видоизменением основной структуры соединения бензольного кольца и гидроксильной группы, базовая комбинация которой и представляет собой фенол. Формула этого вещества в химической записи выглядит как C 6 H 5 OH.
Различные орто-, мета-, паракрезолы являются лишь видоизменением основной структуры соединения бензольного кольца и гидроксильной группы, базовая комбинация которой и представляет собой фенол. Формула этого вещества в химической записи выглядит как C 6 H 5 OH.
Физические свойства фенола
Визуально фенол представляет собой твердые бесцветные кристаллы. На открытом воздухе они окисляются, придавая веществу характерный розовый оттенок. При нормальных условиях фенол довольно плохо растворяется в воде, но с повышением температуры до 70 о этот показатель резко возрастает. В щелочных растворах это вещество растворимо в любых количествах и при любых температурах.
Эти свойства сохраняются и в других соединениях, основным компонентом которых являются фенолы.
Химические свойства
Уникальные свойства фенола объясняются его внутренней структурой. В молекуле этого химического вещества р-орбиталь кислорода образует единую п-систему с бензольным кольцом. Такое плотное взаимодействие повышает электронную плотность ароматического кольца и понижает этот показатель у атома кислорода. При этом полярность связей гидроксогруппы значительно увеличивается, и водород, входящий в ее состав, легко замещается любым щелочным металлом. Так образуются различные феноляты. Эти соединения не разлагаются водой, как алкоголяты, но их растворы очень похожи на соли сильных оснований и слабых кислот, поэтому они имеют достаточно выраженную щелочную реакцию. Феноляты взаимодействуют с различными кислотами, в результате реакции восстанавливаются фенолы. Химические свойства этого соединения позволяют ему взаимодействовать с кислотами, образуя при этом сложные эфиры. Например, взаимодействие фенола и уксусной кислоты приводит к образованию финилового эфира (фениацетата).
При этом полярность связей гидроксогруппы значительно увеличивается, и водород, входящий в ее состав, легко замещается любым щелочным металлом. Так образуются различные феноляты. Эти соединения не разлагаются водой, как алкоголяты, но их растворы очень похожи на соли сильных оснований и слабых кислот, поэтому они имеют достаточно выраженную щелочную реакцию. Феноляты взаимодействуют с различными кислотами, в результате реакции восстанавливаются фенолы. Химические свойства этого соединения позволяют ему взаимодействовать с кислотами, образуя при этом сложные эфиры. Например, взаимодействие фенола и уксусной кислоты приводит к образованию финилового эфира (фениацетата).
Широко известна реакция нитрирования, в которой под воздействием 20% азотной кислоты фенол образует смесь пара- и ортонитрофенолов. Если воздействовать на фенол концентрированной азотной кислотой, то получается 2,4,6-тринитрофенол, который иногда называют пикриновой кислотой.
Фенол в природе
Как самостоятельное вещество фенол в природе содержится в каменноугольной смоле и в отдельных сортах нефти. Но для промышленных нужд это количество не играет никакой роли. Поэтому получение фенола искусственным способом стало приоритетной задачей для многих поколений ученых. К счастью, эту проблему удалось разрешить и получить в итоге искусственный фенол.
Но для промышленных нужд это количество не играет никакой роли. Поэтому получение фенола искусственным способом стало приоритетной задачей для многих поколений ученых. К счастью, эту проблему удалось разрешить и получить в итоге искусственный фенол.
Свойства, получение
Применение различных галогенов позволяет получать феноляты, из которых при дальнейшей обработке образуется бензол. Например, нагревание гидроксида натрия и хлорбензола позволяет получить натрия фенолят, который при воздействии кислоты распадается на соль, воду и фенол. Формула такой реакции приведена здесь:
С 6 Н 5 -CI + 2NaOH -> С 6 Н 5 -ONa + NaCl + Н 2 O
Ароматические сульфокислоты также являются источником для получения бензола. Химическая реакция проводится при одновременном плавлении щелочи и сульфокислоты. Как видно из реакции, сначала образуются феноксиды. При обработке сильными кислотами они восстанавливаются до многоатомных фенолов.
Фенол в промышленности
В теории, получение фенола самым простым и многообещающим способом выглядит таким образом: при помощи катализатора бензол окисляют кислородом.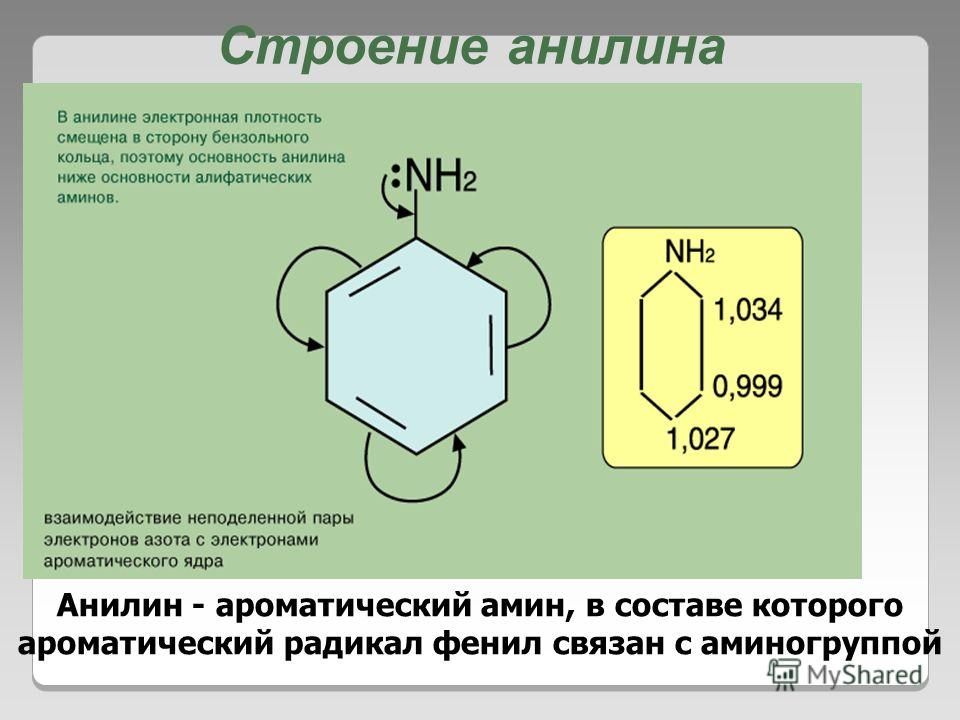 Но до сих пор катализатор для этой реакции так и не был подобран. Поэтому в настоящее время в промышленности используются другие методы.
Но до сих пор катализатор для этой реакции так и не был подобран. Поэтому в настоящее время в промышленности используются другие методы.
Непрерывный промышленный способ получения фенола состоит во взаимодействии хлорбензола и 7% раствора едкого натра. Полученную смесь пропускают через полуторакилометровую систему труб, нагретых до температуры в 300 С. Под воздействием температуры и поддерживаемого высокого давления исходные вещества вступают в реакцию, в результате которой получат 2,4-динитрофенол и другие продукты.
Не так давно был разработан промышленный способ получения фенолсодержащих веществ кумольным методом. Этот процесс состоит из двух этапов. Сначала из бензола получают изопропилбензол (кумол). Для этого бензол алкируют с помощью пропилена. Реакция выглядит следующим образом:
После этого кумол окисляют кислородом. На выходе второй реакции получают фенол и другой важный продукт — ацетон.
Получение фенола в промышленных масштабах возможно из толуола. Для этого толуол окисляется на кислороде, содержащемся в воздухе. Реакция протекает в присутствии катализатора.
Для этого толуол окисляется на кислороде, содержащемся в воздухе. Реакция протекает в присутствии катализатора.
Примеры фенолов
Ближайшие гомологи фенолов называются крезолами.
Существуют три разновидности крезолов. Мета-крезол при нормальных условиях представляет собой жидкость, пара-крезол и орто-крезол — твердые вещества. Все крезолы плохо растворяются в воде, а по своим химическим свойствами они почти аналогичны фенолу. В естественном виде крезолы содержатся в каменноугольной смоле, в промышленности их применяют при производстве красителей, некоторых видов пластмасс.
Примерами двухатомных фенолов могут служить пара-, орто- и мета-гидробензолы. Все они представляют собой твердые вещества, легко растворимые в воде.
Единственный представитель трехатомного фенола — пирогаллол (1,2,3-тригидроксибензол). Его формула представлена ниже.
Пирогаллол является довольно сильным восстановителем. Он легко окисляется, поэтому его используют для получения очищенных от кислорода газов. Это вещество хорошо известно фотографам, его используют как проявитель.
Это вещество хорошо известно фотографам, его используют как проявитель.
Произошла ошибка при настройке пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка браузера на прием файлов cookie
Существует множество причин, по которым файл cookie не может быть установлен правильно. Ниже приведены наиболее распространенные причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки браузера, чтобы принять файлы cookie, или спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает, хотите ли вы принимать файлы cookie, и вы отказались. Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файл cookie.
- Ваш браузер не поддерживает файлы cookie. Попробуйте другой браузер, если вы подозреваете это.
- Дата на вашем компьютере в прошлом. Если часы вашего компьютера показывают дату до 1 января 1970 г., браузер автоматически забудет файл cookie. Чтобы это исправить, установите правильное время и дату на своем компьютере.
- Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу. Предоставить доступ без файлов cookie потребует от сайта создания нового сеанса для каждой посещаемой вами страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в файле cookie; никакая другая информация не фиксируется.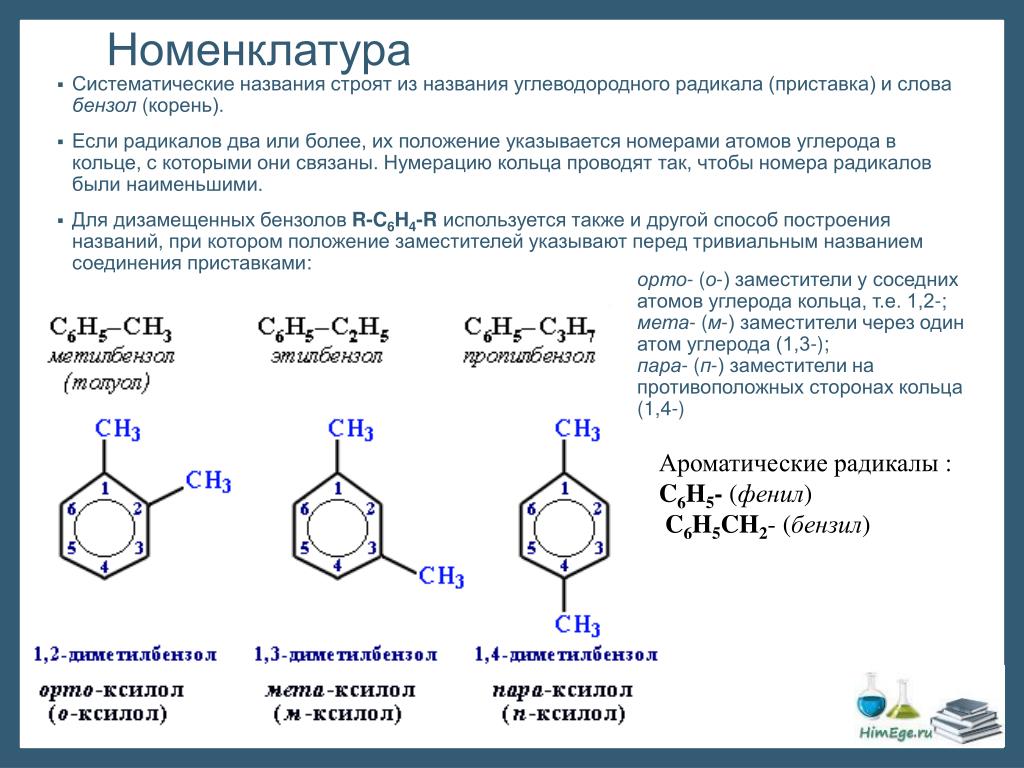
Как правило, в файле cookie может храниться только та информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта. Например, сайт не может определить ваше имя электронной почты, если вы не решите ввести его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступ к остальной части вашего компьютера, и только сайт, создавший файл cookie, может его прочитать.
Реакционная способность радикалов ОН по отношению к фенольным загрязнителям в воде: температурная зависимость констант скорости и новое понимание образования аддукта [ОН-фенол]˙
Известно, что замещенные фенолы легко реагируют с гидроксильным радикалом (OH˙), который является наиболее сильным атмосферным окислителем и также чаще всего используется в усовершенствованных процессах окисления (АОП) для очистки сточных вод. Приведены зависящие от температуры (278,15–318,15 K) кинетические константы скорости второго порядка для воднофазных реакций OH˙ с фенолом и четырьмя замещенными фенолами: катехолом, флороглюцином, пирогаллолом и 3-метилкатехолом, причем последние две измерены для первого время.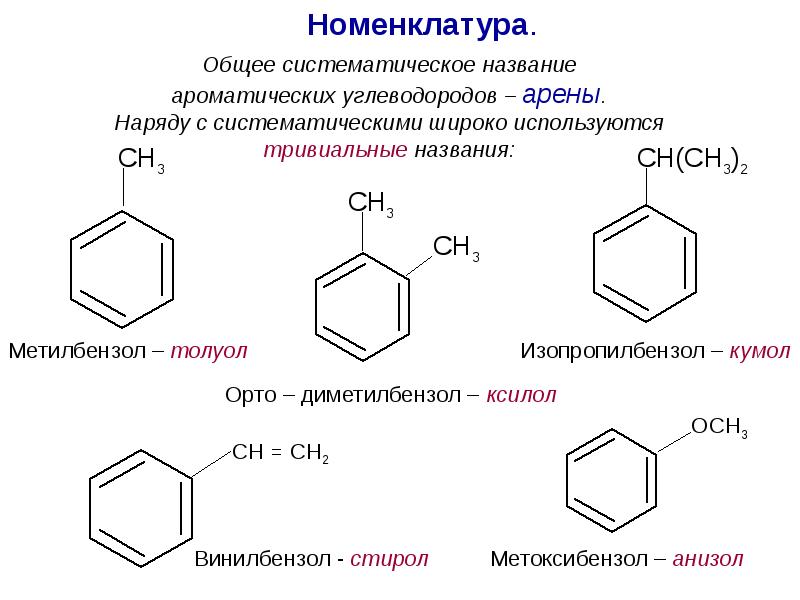 Построенные графики Гаммета для моно- и дизамещенных фенолов имеют потенциал дальнейшего применения для прогнозирования констант скорости реакции других замещенных фенолов при 298,15 К. Это значительно облегчит оптимизацию АОП и улучшит прогностические возможности атмосферных многофазных моделей. в будущем. Более того, теоретические расчеты на уровне M06-2X позволили продвинуться вперед в понимании основного механизма, а именно присоединения , т.е. OH˙ к ароматическому кольцу.Мы показываем, что положение заместителей в ароматическом кольце важно для образования аддукта [ОН-фенол]˙, что подтверждается экспериментом и теоретическими расчетами. Смежные и несмежные заместители донора / акцептора электронов по-разному влияют на взаимодействие между энергией активации и энтропией. Мы также показываем, что явная сольватация должна учитываться в теоретических моделях, чтобы явно описать образование переходного состояния.
Построенные графики Гаммета для моно- и дизамещенных фенолов имеют потенциал дальнейшего применения для прогнозирования констант скорости реакции других замещенных фенолов при 298,15 К. Это значительно облегчит оптимизацию АОП и улучшит прогностические возможности атмосферных многофазных моделей. в будущем. Более того, теоретические расчеты на уровне M06-2X позволили продвинуться вперед в понимании основного механизма, а именно присоединения , т.е. OH˙ к ароматическому кольцу.Мы показываем, что положение заместителей в ароматическом кольце важно для образования аддукта [ОН-фенол]˙, что подтверждается экспериментом и теоретическими расчетами. Смежные и несмежные заместители донора / акцептора электронов по-разному влияют на взаимодействие между энергией активации и энтропией. Мы также показываем, что явная сольватация должна учитываться в теоретических моделях, чтобы явно описать образование переходного состояния.
У вас есть доступ к этой статье
Подождите, пока мы загрузим ваш контент. {-1})$, что указывает на то, что в этих случаях реакция протекает через перенос электрона, а не присоединение.
Информация о журнале
{-1})$, что указывает на то, что в этих случаях реакция протекает через перенос электрона, а не присоединение.
Информация о журнале
Radiation Research публикует статьи, посвященные радиационным эффектам и смежным темам в области физики, химии, биологии и медицины, включая эпидемиологию и трансляционные исследования. Термин «излучение» используется в самом широком смысле и включает, в частности, ионизирующий и ультрафиолетовый, видимый и инфракрасный свет, а также микроволны, ультразвук и тепло.Связанные предметы включают (но не ограничиваются ими) исследования с химическими агентами, способствующие пониманию эффектов радиации, изотопных методов, методов и приборов дозиметрии.
Информация об издателе
Общество радиационных исследований преследует три цели:
самым широким образом поощрять развитие радиационных исследований во всех областях естественных наук;
Содействовать совместным исследованиям между дисциплинами физики, химии, биологии и медицины в изучении
свойства и эффекты радиации;
Способствовать распространению знаний в этих и смежных областях посредством публикаций, встреч и образовательных симпозиумов.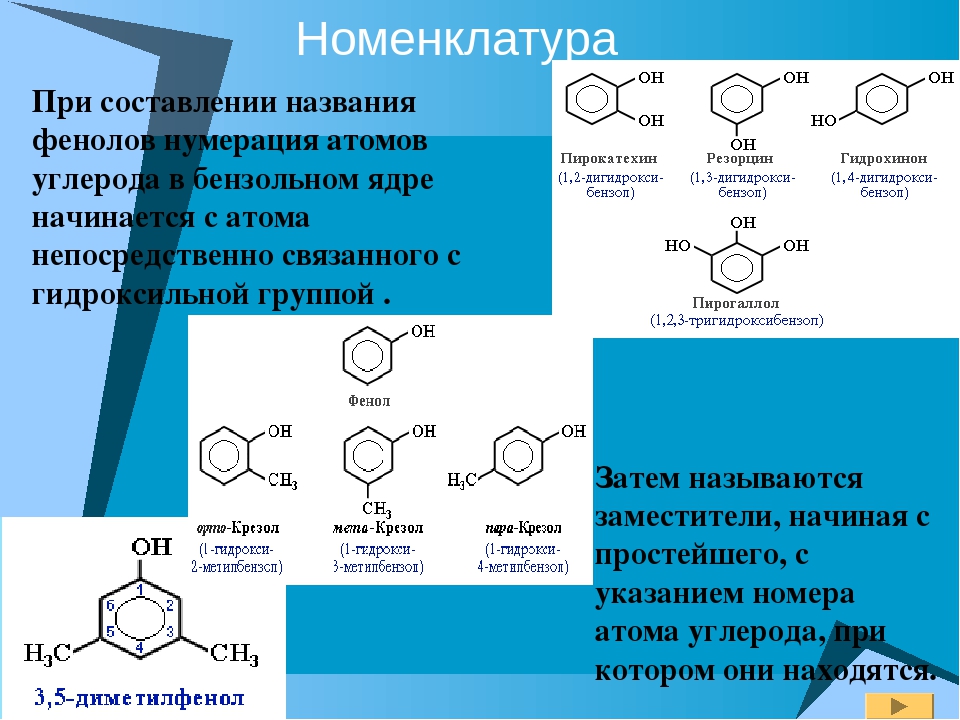
1.32: Окисление боковой цепи, фенолы, ариламины
Карбкатионы, стабилизированные резонансом
Начнем с добавления HCl к алкену, в котором один из атомов углерода алкена непосредственно присоединен к бензольному кольцу.
В этом примере каждый из атомов углерода алкена имеет одинаковое количество атомов водорода, поэтому прямое применение правила Марковникова затруднено. Мы находим, что мы должны выйти за рамки правила Марковникова и объяснить его относительной стабильностью двух карбокатионов, которые могут образоваться.Вот два карбокатиона, которые образуются при атаке H + на алкен:
Какой из этих карбокатионов имеет меньшую энергию? Будет ли фенильная группа отдавать электроны более эффективно, чем метильная группа? Ключевой момент, на который следует обратить внимание при ответе на эти вопросы, заключается в том, что пи-электроны бензольного кольца непосредственно примыкают к углероду карбокатиона в структуре слева.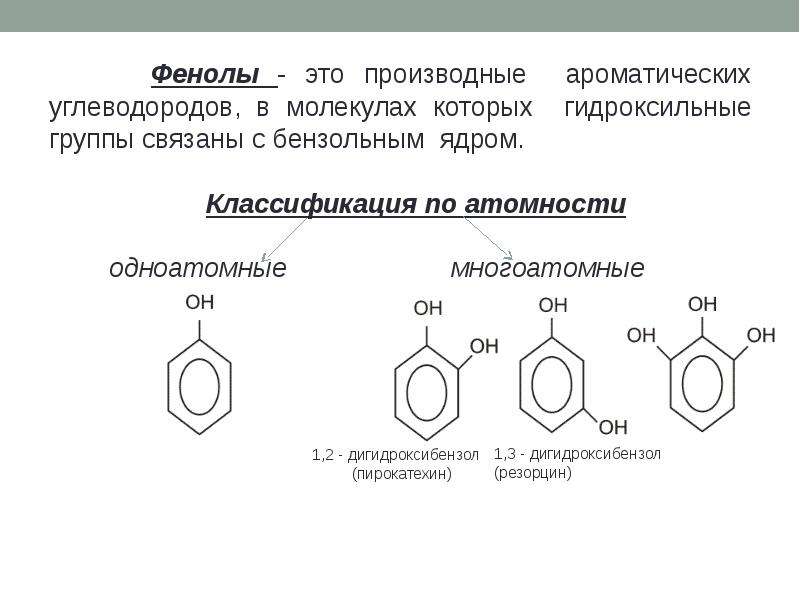 Это напоминает ситуацию, которую мы наблюдали при присоединении HCl к 1,3-бутадиену, где мы столкнулись с резонансной стабилизацией карбокатиона пи-электронами соседней двойной связи.Здесь происходит та же ситуация, и мы можем символизировать стабилизацию резонанса следующими резонансными структурами.
Это напоминает ситуацию, которую мы наблюдали при присоединении HCl к 1,3-бутадиену, где мы столкнулись с резонансной стабилизацией карбокатиона пи-электронами соседней двойной связи.Здесь происходит та же ситуация, и мы можем символизировать стабилизацию резонанса следующими резонансными структурами.
Эффект этого резонанса заключается в том, чтобы сделать карбокатион более стабильным, когда заряд и недостаток электронов расположены на углероде, который непосредственно связан с фенильной группой. Такой углерод называется «бензильным» углеродом. Поскольку при присоединении простой алкильной группы к карбокатионному углероду такая стабилизация резонанса невозможна, бензильный карбокатион более стабилен, образуется быстрее, и продукт, образующийся из него, является единственным, который мы видим.
На этом фоне мы можем быстро решить, какой из двух приведенных ниже алкилгалогенидов будет реагировать быстрее по механизму S N 1.
Более реакционноспособный алкилгалогенид дает более стабильный карбокатион. Более стабильный карбокатион (поскольку оба будут вторичными карбокатионами) — это тот, в котором возможен бензильный резонанс. (Резонансные структуры приведены выше.) Только алкилгалогенид справа образует карбокатион, в котором углерод карбокатиона непосредственно присоединен к фенильной группе.Следовательно, мы понимаем, почему алкилгалогенид справа более реакционноспособен.
Более стабильный карбокатион (поскольку оба будут вторичными карбокатионами) — это тот, в котором возможен бензильный резонанс. (Резонансные структуры приведены выше.) Только алкилгалогенид справа образует карбокатион, в котором углерод карбокатиона непосредственно присоединен к фенильной группе.Следовательно, мы понимаем, почему алкилгалогенид справа более реакционноспособен.
Основания, стабилизированные резонансом
Мы видели, что бензильные карбокатионы стабилизируются резонансом. Что произойдет, если атом, непосредственно присоединенный к бензольному кольцу, имеет неподеленную пару электронов — другими словами, он богат электронами, а не беден электронами. Мы можем видеть эффекты такого расположения в кислотно-основных свойствах фенолов и ароматических аминов.
Фенол — это спирт, R-группа которого представляет собой фенильную группу.Важно, чтобы кислородное и бензольное кольцо были непосредственно связаны друг с другом. Если между ними находится тетраэдрический углерод, то соединение представляет собой не фенол, а спирт.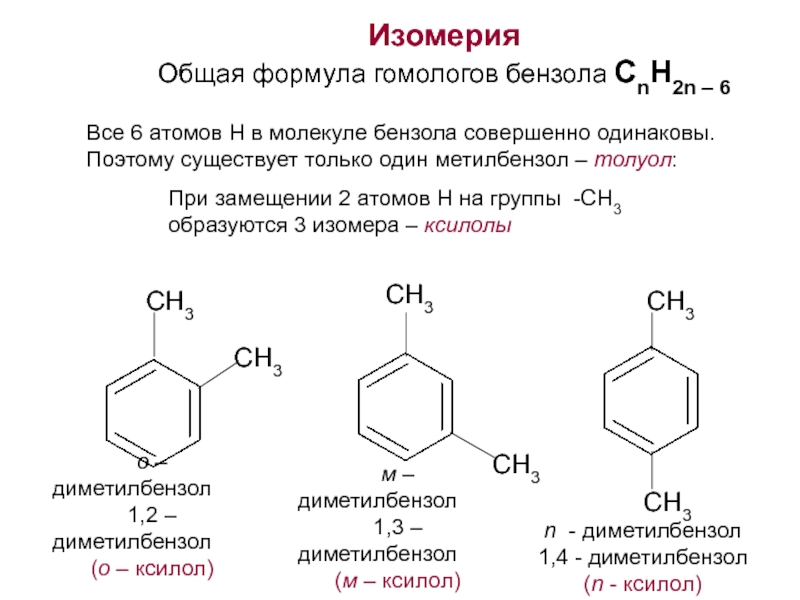
Важное различие между фенолами и спиртами заключается в их кислотности. Напомним, что спирты, как и вода, имеют p K a около 16. Фенолы, напротив, обычно имеют p K a около 10, что делает их значительно более сильными кислотами, чем спирты.Поскольку более сильная кислота имеет более слабое сопряженное основание, нам нужно спросить, почему феноксид (сопряженное основание фенола) слабее алкоксида (сопряженное основание спирта).
Поскольку именно неподеленная электронная пара делает любое из оснований основанием, мы можем спросить, почему неподеленная электронная пара на феноксид-ионе менее доступна для связи с протоном, чем неподеленная электронная пара на алкоксид-ионе. Опять же, причина находится в резонансе.
Резонансные структуры, которые улучшают описание феноксид-иона, показывают, что электронная пара на кислороде частично перемещается в положение пи-связи между кислородом и фенильным углеродом, к которому она присоединена.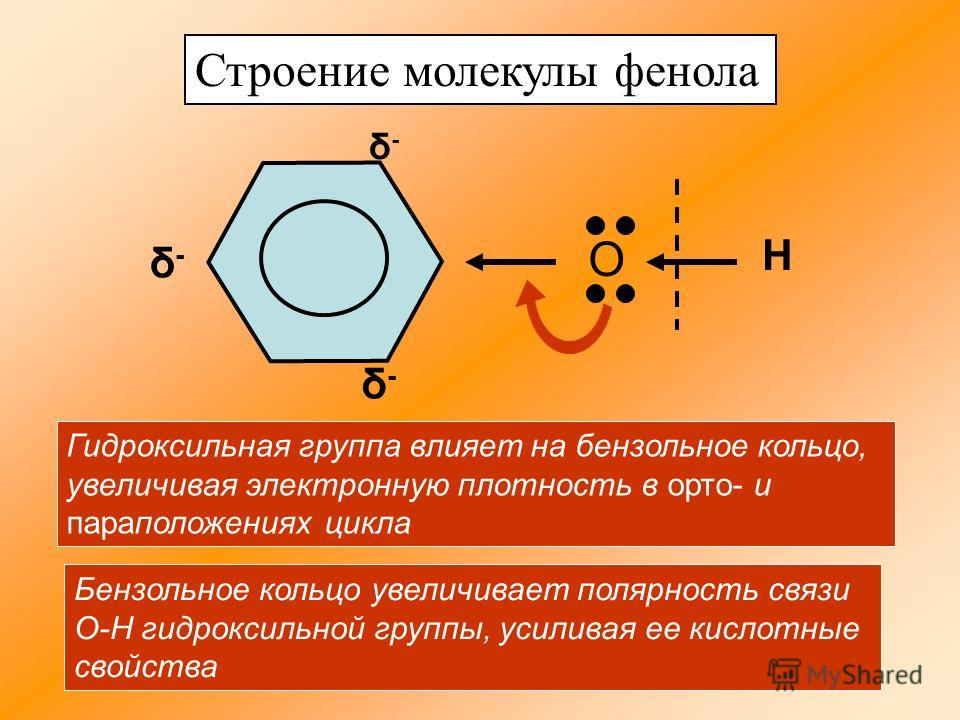 В связи с этим электронная пара на феноксид-ионе менее доступна для присоединения к протону, а феноксид-ион является более слабым основанием, чем алкоксид-ион, в котором такой резонанс невозможен.
В связи с этим электронная пара на феноксид-ионе менее доступна для присоединения к протону, а феноксид-ион является более слабым основанием, чем алкоксид-ион, в котором такой резонанс невозможен.
Такая же ситуация возникает, когда азот аминогруппы напрямую связан с ароматическим кольцом.
Мы видим, что ион аммония, в котором азот непосредственно присоединен к фенильной группе, является более сильной кислотой, чем тот, в котором азот присоединен к sp 3 гибридизованному атому углерода.Это означает, что сопряженное основание, ариламин, в котором азот присоединен к фенильной группе, также слабее, чем его аналог алкиламина. Это понимается под резонансом между неподеленной электронной парой азота и ароматическим кольцом, возможным только в том случае, если связь прямая, что делает неподеленную электронную пару менее доступной для использования в качестве основания.
Резонансные возможности ароматической группы позволяют ей служить либо заместителем, отдающим электроны, когда присоединенный атом нуждается в электронах (карбокатион), либо заместителем, отдающим электроны, когда присоединенный атом имеет неподеленную пару (кислород или азот).
Окисление боковой цепи
Существует еще одна реакция, которая происходит на атоме, непосредственно присоединенном к ароматическому кольцу. Это известно как «окисление боковой цепи». Когда соединение, имеющее алкильную группу, непосредственно присоединенную к арильной группе, обрабатывается сильным окислителем, таким как хромовая кислота, бензильный углерод окисляется до группы карбоновой кислоты, которая остается присоединенной к арильной группе. Любые другие углерод-углеродные связи разорваны. Есть еще одно ограничение — к бензиловому углероду должен быть присоединен водород.
Механизм этой реакции неясен, но тот факт, что она требует наличия бензильной связи C-H, предполагает, что разрыв этой связи необходим. Любое промежуточное соединение, которое может образоваться при разрыве этой связи, будет стабилизировано за счет резонанса с арильной группой, что объясняет специфичность атаки по бензильному положению. Такие реакции также происходят в биологическом контексте.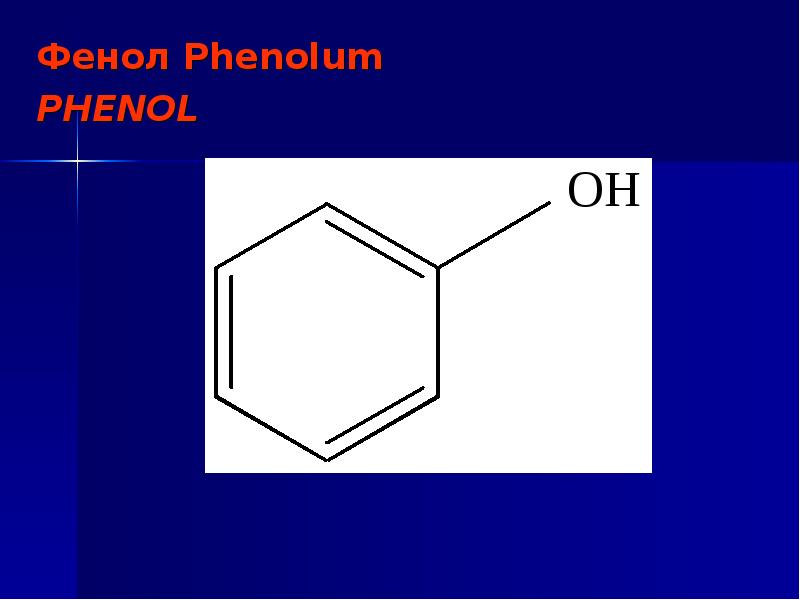 Ферменты окисляют боковые алкильные цепи ароматических колец, делая такие соединения достаточно растворимыми, чтобы их можно было удалить.
Ферменты окисляют боковые алкильные цепи ароматических колец, делая такие соединения достаточно растворимыми, чтобы их можно было удалить.
Ионы диазония
Когда первичные ариламины реагируют с азотистой кислотой (HNO 2 , образующейся из нитрита натрия (NaNO 2 и HCl), происходит реакция, в результате которой образуется новая тройная связь азот-азот. Мы не будем касаться механизма, но продукт (называемый ионом диазония) является ценным синтетическим промежуточным продуктом.
Ион диазония редко выделяется (ионы диазония могут быть взрывоопасными), поэтому вместо этого он используется в растворе, в котором он сделан.Группа N 2 может быть заменена множеством других групп, а также сама может быть электрофилом по отношению к достаточно активированному ароматическому кольцу. Возможности представлены в следующей таблице:
Первые четыре реакции на этой диаграмме являются примерами замены группы N 2 нуклеофилом. Эти реакции обеспечивают способ осуществления нуклеофильных замещений на углероде ароматического кольца, которые невозможны по обычным механизмам S N 1 и S N 2 .Большая стабильность молекулярного азота (N 2 ) делает его очень хорошей уходящей группой, но механизмы этих реакций непросты.
Эти реакции обеспечивают способ осуществления нуклеофильных замещений на углероде ароматического кольца, которые невозможны по обычным механизмам S N 1 и S N 2 .Большая стабильность молекулярного азота (N 2 ) делает его очень хорошей уходящей группой, но механизмы этих реакций непросты.
Последняя реакция в таблице — это реакция, в которой группа диазония действует как электрофил. Это не особенно хороший электрофил, поэтому в качестве мишени ему требуется реактивное кольцо. Такое кольцо должно иметь сильную электронодонорную группу, такую как OH или NH 2 , чтобы увеличить его реакционную способность в механизме электрофильного ароматического замещения.
Продукт электрофильного сочетания иона диазония с реакционноспособным ароматическим кольцом включает двойную связь азот-азот. Эти соединения часто имеют интенсивную окраску и используются в качестве красителей. Могут быть включены различные другие функциональные группы, часто несущие ионные заряды, чтобы они плотнее прилипали к ткани. Для обозначения этих соединений часто используются такие термины, как анилиновые красители или азокрасители.
Для обозначения этих соединений часто используются такие термины, как анилиновые красители или азокрасители.
Соли диазония являются универсальными синтетическими промежуточными материалами, и мы знаем, что они сделаны из анилина или подобных молекул.Откуда берутся анилин и его аналоги? Наиболее распространенный способ — восстановление нитробензола или замещенных нитробензолов. Обычно восстановителем является олово (в лаборатории) или железо (в производственной практике), используемые в водном растворе HCl. Поскольку нитрование является широко используемым путем получения нитробензола, обычная последовательность производства анилина такова:
Использование NaOH на последнем этапе заключается в нейтрализации HCl таким образом, чтобы основной амин (который в противном случае был бы в форме его сопряженного кислотного иона аммония) был достаточно неполярным, чтобы его можно было экстрагировать из воды.
Синтез фенолов
В процессе Доу хлорбензол реагирует с разбавленным гидроксидом натрия при 300°C и давлении 3000 фунтов на квадратный дюйм.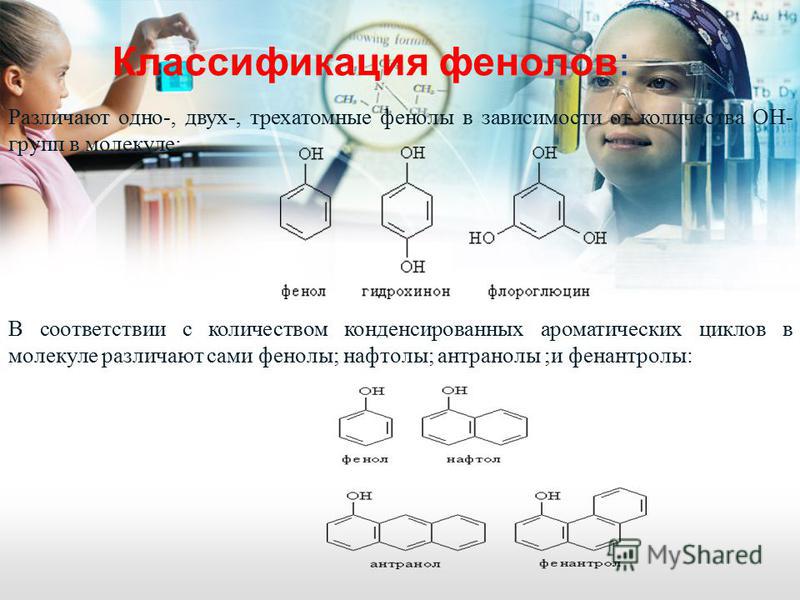 Следующий рисунок иллюстрирует процесс Доу.
Следующий рисунок иллюстрирует процесс Доу.
Окисление кумола (изопропилбензола) воздухом приводит к производству как фенола, так и ацетона, как показано на следующем рисунке. Механизмы образования и разложения гидропероксида кумола требуют более подробного рассмотрения, которое представлено после рисунка.
Образование гидропероксидов кумола. Образование гидропероксида происходит в результате свободнорадикальной цепной реакции. Радикальный инициатор отделяет безводородный радикал от молекулы, создавая третичный свободный радикал. Создание третичного свободного радикала является начальным этапом реакции.
На следующем этапе свободный радикал притягивается к молекуле кислорода. Это притяжение производит свободный радикал гидропероксида.
Наконец, свободный радикал гидропероксида отрывает свободный радикал водорода от второй молекулы кумола с образованием гидропероксида кумола и нового третичного свободного радикала.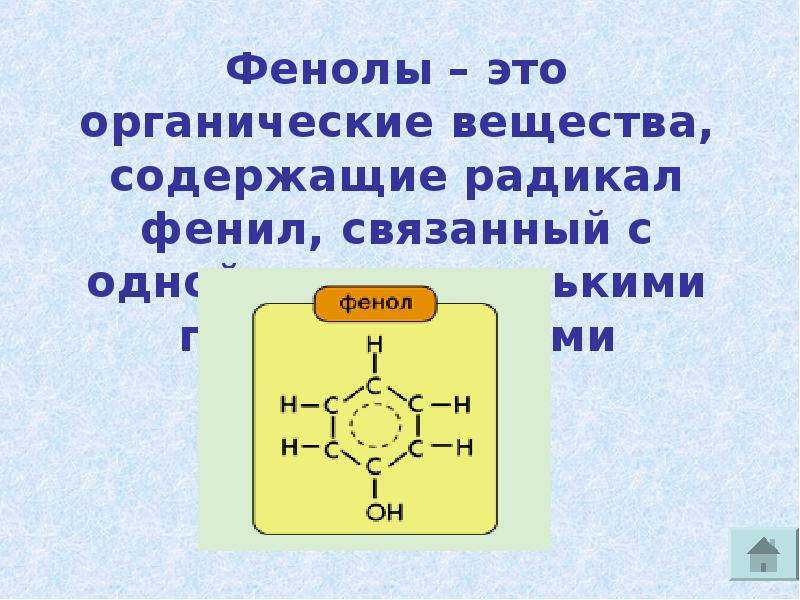
Разложение гидропероксида кумола. Деструкция гидропероксида кумола протекает по карбокатионному механизму. На первом этапе пара электронов кислорода «гидроксильной группы» гидропероксида притягивается к протону молекулы H 3 O + , образуя ион оксония.
Затем ион оксония стабилизируется, когда положительно заряженный кислород уходит в молекулу воды. Эта потеря молекулы воды приводит к образованию нового иона оксония.
Сдвиг фенид-иона к атому кислорода (который создает третичный карбокатион) стабилизирует положительно заряженный кислород. (Фенид-ион представляет собой фенильную группу с электронной связывающей парой, доступной для образования новой связи с кольцом.)
Карбкатион стабилизируется кислотно-щелочной реакцией с молекулой воды, что приводит к образованию иона оксония.
Потеря протона стабилизирует ион оксония.
Затем протон подхватывается кислородом эфира в кислотно-щелочной реакции с образованием нового иона оксония.
Положительно заряженный эфирный кислород притягивает к себе электроны в связи кислород-углерод, тем самым делокализуя заряд обоих атомов. Частичный положительный заряд на углероде притягивает несвязывающую электронную пару от кислорода группы ОН, позволяя электронам в исходной кислородно-углеродной связи высвобождаться обратно к более электроотрицательному атому кислорода.
Наконец, протон теряется из протонированной молекулы ацетона, что приводит к образованию ацетона.
Селективное превращение лигнина в фенол путем прямого превращения связей Csp2–Csp3 и C–O . Ожидается, что мировой рынок фенола будет постоянно расти из-за его коммерческой жизнеспособности и растущего спроса на его широкое использование в качестве промежуточного органического химического вещества и сырья для производства различных промышленных продуктов, в частности, фенольной смолы, бисфенола А, нейлона, циклогексанола, эпоксидной смолы. , поликарбонат, фармацевтический и т.
 д.( 3 ). В промышленности фенол в основном получают из бензола путем многостадийного и непрямого синтеза, а именно кумольного процесса, который имеет ряд недостатков, таких как сложные и тяжелые условия, высокое потребление энергии и ископаемого сырья, серьезное загрязнение окружающей среды ( 2 , 4 ). Хотя прямое гидроксилирование ароматического кольца ( 5 , 6 ) считается простым и экологически безопасным процессом производства фенола, этот путь по-прежнему зависит от ископаемого бензола и ограничен переокислением фенола ( 4 ).Таким образом, желательна более эффективная и устойчивая стратегия, такая как использование биомассы в качестве сырья для экономичного производства фенола, которая может освободить нас от зависимости от ископаемых ресурсов и имеет большое промышленное и социальное значение ( 7 ). основной компонент лигноцеллюлозной биомассы, лигнин является наиболее распространенным возобновляемым источником, состоящим из ароматических единиц в природе, что делает упор на стратегии, которые получают ароматические химические вещества с добавленной стоимостью из биомассы ( 8 – 11 ).
д.( 3 ). В промышленности фенол в основном получают из бензола путем многостадийного и непрямого синтеза, а именно кумольного процесса, который имеет ряд недостатков, таких как сложные и тяжелые условия, высокое потребление энергии и ископаемого сырья, серьезное загрязнение окружающей среды ( 2 , 4 ). Хотя прямое гидроксилирование ароматического кольца ( 5 , 6 ) считается простым и экологически безопасным процессом производства фенола, этот путь по-прежнему зависит от ископаемого бензола и ограничен переокислением фенола ( 4 ).Таким образом, желательна более эффективная и устойчивая стратегия, такая как использование биомассы в качестве сырья для экономичного производства фенола, которая может освободить нас от зависимости от ископаемых ресурсов и имеет большое промышленное и социальное значение ( 7 ). основной компонент лигноцеллюлозной биомассы, лигнин является наиболее распространенным возобновляемым источником, состоящим из ароматических единиц в природе, что делает упор на стратегии, которые получают ароматические химические вещества с добавленной стоимостью из биомассы ( 8 – 11 ).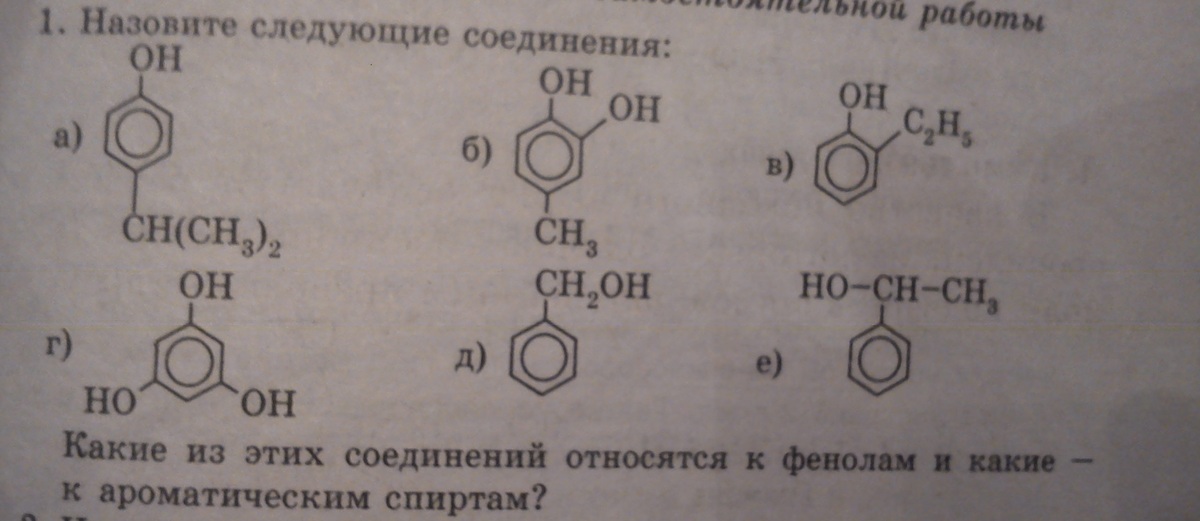 Синтез фенола с использованием лигнина является многообещающим, но сложным путем. В последнее время достигнуты заметные успехи. Селс и его коллеги ( 12 ) разработали интегрированный процесс биопереработки, который превращает 78 мас.% (вес.%) березы в ксилохимические вещества. В этом процессе восстановительное каталитическое фракционирование древесины первоначально дает углеводную целлюлозу, пригодную для производства биоэтанола и лигнинового масла. После экстракции смесь фенольных мономеров в лигниновом масле каталитически направляют в 20 мас.% фенола (в расчете на массу лигнина) путем последовательной газофазной гидрообработки и деалкилирования (рис.1А, и подробный процесс, показанный на фиг. С1А). Ван и его коллеги ( 13 ) предложили многоступенчатую методологию производства фенола из лигнина (рис. 1B и подробная методология, показанная на рис. S1B). На первой стадии структура (Ar)C α (OH)–C β в лигнине окисляется до промежуточного соединения C α (=O)–C β .
Синтез фенола с использованием лигнина является многообещающим, но сложным путем. В последнее время достигнуты заметные успехи. Селс и его коллеги ( 12 ) разработали интегрированный процесс биопереработки, который превращает 78 мас.% (вес.%) березы в ксилохимические вещества. В этом процессе восстановительное каталитическое фракционирование древесины первоначально дает углеводную целлюлозу, пригодную для производства биоэтанола и лигнинового масла. После экстракции смесь фенольных мономеров в лигниновом масле каталитически направляют в 20 мас.% фенола (в расчете на массу лигнина) путем последовательной газофазной гидрообработки и деалкилирования (рис.1А, и подробный процесс, показанный на фиг. С1А). Ван и его коллеги ( 13 ) предложили многоступенчатую методологию производства фенола из лигнина (рис. 1B и подробная методология, показанная на рис. S1B). На первой стадии структура (Ar)C α (OH)–C β в лигнине окисляется до промежуточного соединения C α (=O)–C β . На второй стадии окислительно расщепляется связь (Ar)C α (=O)–C β с образованием структуры (Ar)C α (=O)–OH. Наконец, арильная связь C–C α (C sp2 –C sp3 ) разрушается посредством декарбоксилирования.Вместе с гидрогенолизом алифатической связи β-углерод-кислород (C β –O) стратегия многостадийного окисления может дать смесь ароматических соединений и фенола с селективностью по фенолу 60% ( 13 ).
На второй стадии окислительно расщепляется связь (Ar)C α (=O)–C β с образованием структуры (Ar)C α (=O)–OH. Наконец, арильная связь C–C α (C sp2 –C sp3 ) разрушается посредством декарбоксилирования.Вместе с гидрогенолизом алифатической связи β-углерод-кислород (C β –O) стратегия многостадийного окисления может дать смесь ароматических соединений и фенола с селективностью по фенолу 60% ( 13 ). Рис. 1 Устойчивые пути производства фенола.
( A ) Комплексный процесс биоочистки для производства фенола из древесины ( 12 ). ( B ) Путь многостадийного окисления-гидрогенолиза фенола из лигнина, C α и C β представляют собой алифатические положения α-C и β-C ( 13 ).( C ) Маршрут этой работы по получению фенола из лигнина путем сочетания прямой деконструкции связи C sp2 –C sp3 и гидролиза алифатической связи C β –O в H ( p -гидроксифенл ) структура; C sp2 –C sp3 представляет собой арил C–C α связи. Разработка нового способа получения фенола из лигнина по-прежнему весьма желательна, хотя сообщалось о некоторых интересных работах. Мы можем предположить из репрезентативного H ( p -гидроксифенил) структуры в древесных и травянистых растениях (рис.S2), что фенол может быть эффективно получен из лигнина прямым разрушением связей C sp2 –C sp3 и алифатических связей C β –O, как показано на рис. 1C. До сих пор были проведены обширные исследования расщепления связи C-O в лигнине, и алкилфенолы могут быть получены с неразорванными связями C sp2 – C sp3 ( 14 – 20 ). В этом контексте эффективное расщепление связей С-С является ключом к получению фенола из лигнина.Расщепление связи C sp2 –C sp3 может осуществляться деалкилированием, деалкенилированием и восстановительным расщеплением. Тем не менее, эти существующие стратегии по-прежнему ограничены суровыми условиями реакции ( 12 , 21 – 23 ).
Разработка нового способа получения фенола из лигнина по-прежнему весьма желательна, хотя сообщалось о некоторых интересных работах. Мы можем предположить из репрезентативного H ( p -гидроксифенил) структуры в древесных и травянистых растениях (рис.S2), что фенол может быть эффективно получен из лигнина прямым разрушением связей C sp2 –C sp3 и алифатических связей C β –O, как показано на рис. 1C. До сих пор были проведены обширные исследования расщепления связи C-O в лигнине, и алкилфенолы могут быть получены с неразорванными связями C sp2 – C sp3 ( 14 – 20 ). В этом контексте эффективное расщепление связей С-С является ключом к получению фенола из лигнина.Расщепление связи C sp2 –C sp3 может осуществляться деалкилированием, деалкенилированием и восстановительным расщеплением. Тем не менее, эти существующие стратегии по-прежнему ограничены суровыми условиями реакции ( 12 , 21 – 23 ). Расщепление связей C sp2 – C sp3 обычно происходит при температуре выше 400°C в процессе деалкилирования на обычном биоперерабатывающем заводе, что приводит к побочным реакциям ( 12 , 24 ).Таким образом, разрыв связи С sp2 – С sp3 в мягких условиях необходим. Исторически сложилось так, что мягкое разрушение связи C–C часто осуществлялось путем сочетания этого процесса с одновременным образованием других связей, таких как образование связи C–X (X = C, Si, N, O, S, F…) ( 25 – 30 ). Метод деконструктивной функционализации имеет ограничения в повышении ценности лигнина, поскольку он включает несколько этапов и имеет низкую экономию атомов ( 7 ). С учетом этих ограничений разработка простых и эффективных методов расщепления связи C sp2 –C sp3 , которые облегчают высокоселективное получение фенола из лигнина, остается важной задачей и научной задачей.Здесь мы сообщаем о механистически отличной и функционально краткой стратегии получения фенола из лигнина путем прямого разрушения связи C sp2 –C sp3 и гидролиза алифатической связи C β –O (рис.
Расщепление связей C sp2 – C sp3 обычно происходит при температуре выше 400°C в процессе деалкилирования на обычном биоперерабатывающем заводе, что приводит к побочным реакциям ( 12 , 24 ).Таким образом, разрыв связи С sp2 – С sp3 в мягких условиях необходим. Исторически сложилось так, что мягкое разрушение связи C–C часто осуществлялось путем сочетания этого процесса с одновременным образованием других связей, таких как образование связи C–X (X = C, Si, N, O, S, F…) ( 25 – 30 ). Метод деконструктивной функционализации имеет ограничения в повышении ценности лигнина, поскольку он включает несколько этапов и имеет низкую экономию атомов ( 7 ). С учетом этих ограничений разработка простых и эффективных методов расщепления связи C sp2 –C sp3 , которые облегчают высокоселективное получение фенола из лигнина, остается важной задачей и научной задачей.Здесь мы сообщаем о механистически отличной и функционально краткой стратегии получения фенола из лигнина путем прямого разрушения связи C sp2 –C sp3 и гидролиза алифатической связи C β –O (рис.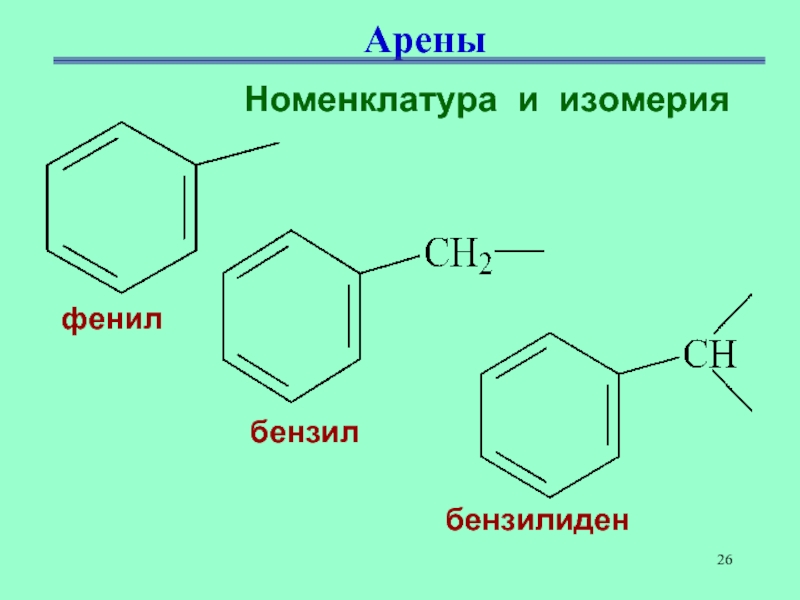 1C). Эта работа предлагает эффективную и экономичную стратегию селективного превращения лигнина в фенол в мягких условиях.
1C). Эта работа предлагает эффективную и экономичную стратегию селективного превращения лигнина в фенол в мягких условиях. РЕЗУЛЬТАТЫ
Как обсуждалось выше, расщепление C sp2 – C sp3 имеет решающее значение для производства фенола из лигнина.Мы начали наши исследования предполагаемого деконструктивного разрыва связи C sp2 –C sp3 с оценки широкого спектра твердокислотных катализаторов, катализаторов на носителе и минеральных кислот с использованием 4-(1-гидроксипропил)фенола ( 1a ) в качестве субстрата (таблица 1 и таблица S1) в воде. После обширного скрининга различных условий мы определили оптимизированные условия, показанные в записи 1, в которых использовался дешевый и безвредный для окружающей среды цеолит HY 30 (Si/Al = 30; рис.S3 и таблица S2) в H 2 O при 180°C в течение 1 часа (рис. S4), что дает выход 94,6% фенола ( 1b ) при полной конверсии 1a (рис. S5). Реакция не протекает без катализатора (опыт 2).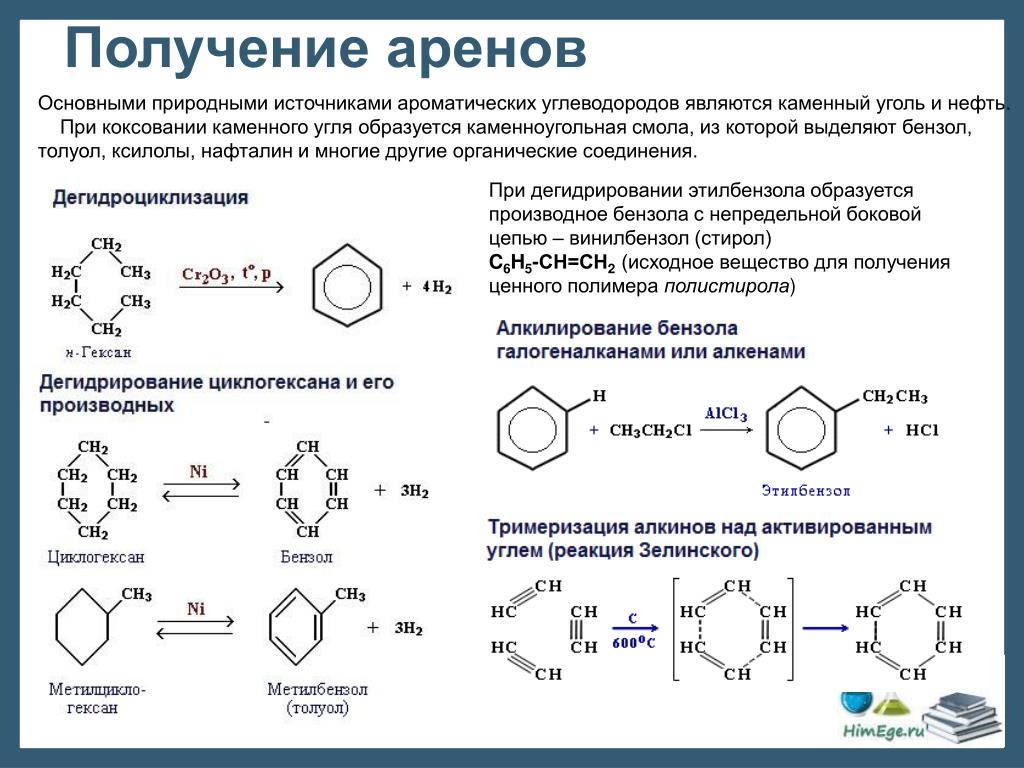 Хорошо известно, что цеолит HY 3 имеет каркас, богатый алюминием ( 31 ). Однако большинство кислотных центров, особенно сильнокислотные центры, встроены глубоко в микропористый каркас, доступ к которому реагентам затруднен, и реакция не может происходить (позиция 3 в таблице 1 для HY 3 ).После изоморфного замещения алюминия каркаса кремнием высококремнистые цеолиты Y, такие как HY 15 , HY 30 и HY 40 , имеют более мезопористую структуру (рис. S3J и таблица S2), что позволяет реагенты свободно диффундируют к открытым сильным кислотным центрам ( 24 ). Однако, в отличие от цеолитов HY 30 и HY 40 , HY 15 имеет менее мезопористую структуру и более низкую концентрацию сильных кислотных центров (таблица S2), что, следовательно, приводит к более низкому выходу 1b в тех же условиях. (запись 3 таблицы 1 для HY 15 ).Цеолиты HY 30 и HY 40 имеют более высокую концентрацию сильнокислотных центров, открытых в мезопористых каналах (таблица S2), что дает им более высокие выходы 1b (записи 1 и 3 таблицы 1).
Хорошо известно, что цеолит HY 3 имеет каркас, богатый алюминием ( 31 ). Однако большинство кислотных центров, особенно сильнокислотные центры, встроены глубоко в микропористый каркас, доступ к которому реагентам затруднен, и реакция не может происходить (позиция 3 в таблице 1 для HY 3 ).После изоморфного замещения алюминия каркаса кремнием высококремнистые цеолиты Y, такие как HY 15 , HY 30 и HY 40 , имеют более мезопористую структуру (рис. S3J и таблица S2), что позволяет реагенты свободно диффундируют к открытым сильным кислотным центрам ( 24 ). Однако, в отличие от цеолитов HY 30 и HY 40 , HY 15 имеет менее мезопористую структуру и более низкую концентрацию сильных кислотных центров (таблица S2), что, следовательно, приводит к более низкому выходу 1b в тех же условиях. (запись 3 таблицы 1 для HY 15 ).Цеолиты HY 30 и HY 40 имеют более высокую концентрацию сильнокислотных центров, открытых в мезопористых каналах (таблица S2), что дает им более высокие выходы 1b (записи 1 и 3 таблицы 1). Однако кислотные центры цеолита HY 40 меньше, чем у HY 30 (таблица S2), и, таким образом, каталитическая активность HY 30 была лучше, чем у цеолита HY 40 . Другие типы цеолитов, такие как ZSM-5, морденит, бета и MCM-41, не могли эффективно стимулировать реакцию в таких условиях (опыты 4-7).Катализаторы на носителе не были активны в реакции (опыты 8-10). Только катализатор SO 4 2− /ZrO 2 может давать небольшое количество продукта (опыт 10). Кроме того, гетерополи и минеральные кислоты не давали 1b (опыты 11 и 12). Приведенные выше результаты показывают, что цеолит HY 30 продемонстрировал исключительную эффективность в качестве катализатора реакции, при которой реакция могла эффективно протекать при умеренной температуре (180°C).
Однако кислотные центры цеолита HY 40 меньше, чем у HY 30 (таблица S2), и, таким образом, каталитическая активность HY 30 была лучше, чем у цеолита HY 40 . Другие типы цеолитов, такие как ZSM-5, морденит, бета и MCM-41, не могли эффективно стимулировать реакцию в таких условиях (опыты 4-7).Катализаторы на носителе не были активны в реакции (опыты 8-10). Только катализатор SO 4 2− /ZrO 2 может давать небольшое количество продукта (опыт 10). Кроме того, гетерополи и минеральные кислоты не давали 1b (опыты 11 и 12). Приведенные выше результаты показывают, что цеолит HY 30 продемонстрировал исключительную эффективность в качестве катализатора реакции, при которой реакция могла эффективно протекать при умеренной температуре (180°C). | вход | Вариация катализатора | Выход /% | |||
|---|---|---|---|---|---|
| 1 | Zeolite HY 30 | 94.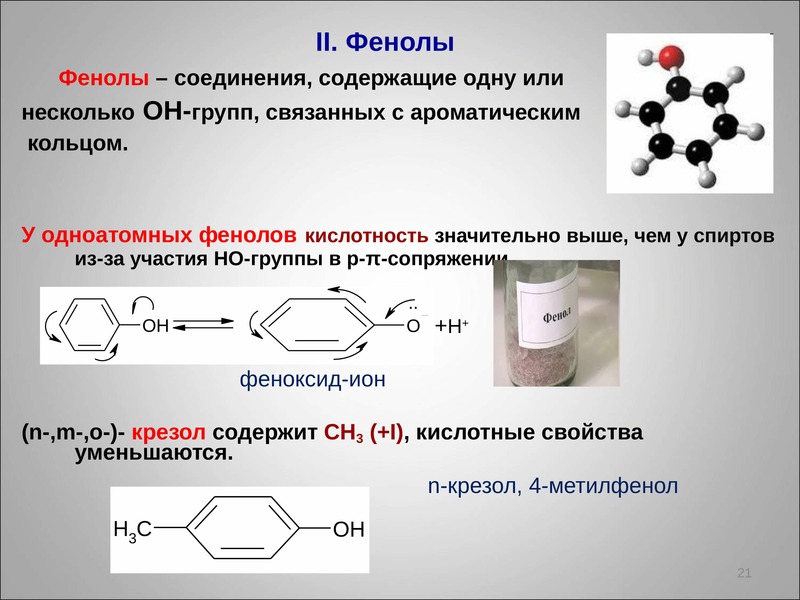 6 6 | |||
| 2 | 2 | 2 | 0 | 0 | |
| 3 | Zeolite HY 3 , HY 15 , и HY 40 | 4 0, 8,7, 51.9 ||||
| 4 | Zeolite ZSM-5 9 , 9 , ZSM-5 30 , ZSM-5 63 , и ZSM-5 235 | 0, 0, 0, 11.3 | |||
| 5 | Zeoolite Mordenite 10 и Mordenite 13 13 | 0, 0 | 0 | 0 | |
| 6 | Zeolite Beta 13 , бета 30 , а бета 50 | 0, 0, 0 | |||
| 7 | 7 | Zeolite MCM-41 | 0 | 0 | 9 |
| 8 | RU / SIO 2 , Ni / Sio 2 , и W / SIO 2 | 0, 0, 0 8 ||||
| 9 | SO42−/ZrO2 и WO 3 / ZrO 2 | 5. 3, 0 3, 0 | |||
| 10 | |||||
| WO 3 / AI 2 / AI 2 O 3 и NB 2 O 5 / AI 2 O 3 | 0, 0 | ||||
| 11 | 11 | гетерополитическая кислота | 0 | 0 | 9 |
| 12 | HCI, H 2 SO 4 , и H 3 PO 4 | 0, 0, 0 |
Таблица 1 Скрининг катализаторов прямого разрушения связи С sp2 – С sp3 .
Стандартные условия: 4-(1-гидроксипропил)фенол ( 1a ; 1 ммоль), катализатор (0,3 г), H 2 O (4,0 мл), 180°C, 1 час, 0,5 МПа Ar, и 800 об/мин.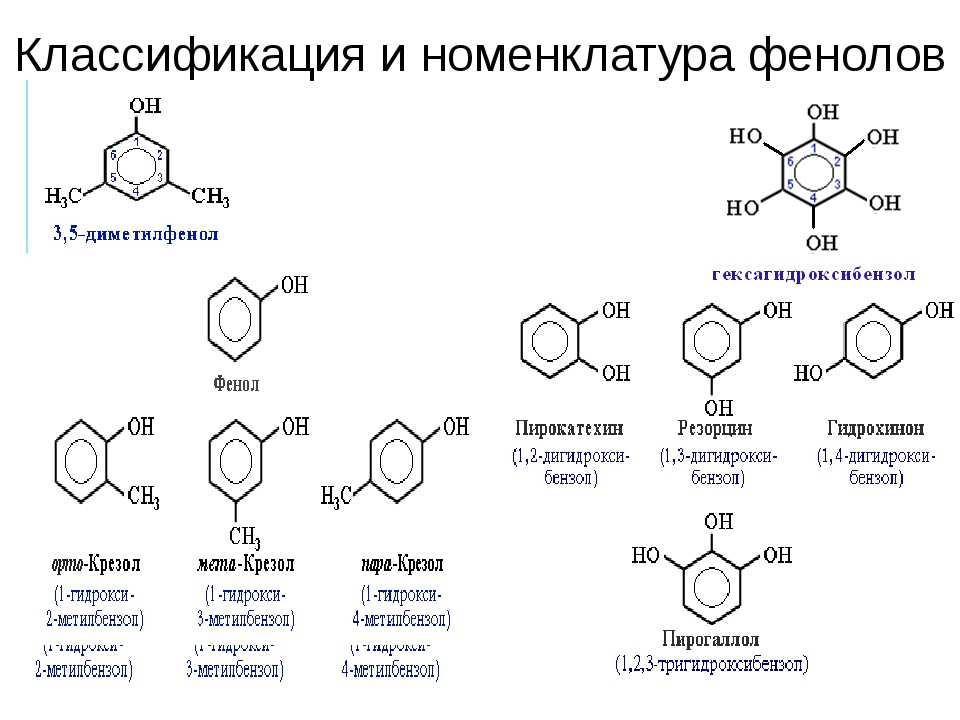 Все результаты реакции представляют собой средние значения трех экспериментов, проведенных параллельно.
Все результаты реакции представляют собой средние значения трех экспериментов, проведенных параллельно.
 Примечательно, что разрыв связей C sp2 –C sp3 в звеньях G и S затруднен из-за стерических и электронных эффектов, что сдерживало образование соответственно 2-метоксифенола и 2,6-диметоксифенола и обеспечивало высокую селективность по фенолу ( 13 ). Нами были также проведены трансформации лигнинов, выделенных из других пород древесины, в том числе из сосны, ивы, кипариса и травянистого растения Phyllostachys pubescens (рис.С8). Выходы фенола составили соответственно 9,0, 6,3, 4,5 и 10,1% (в пересчете на лигнин) при высокой селективности. Чтобы получить чистый фенол, мы провели масштабный эксперимент по трансформации лигнина тополя (рис. S9), в результате которого из 50,0 г лигнина было получено 4,1 г чистого фенола. Выход фенола был несколько снижен по сравнению с результатом газовой хроматографии (ГХ), в основном из-за потерь в процессе разделения. Чтобы дополнительно доказать эффективность катализатора, мы изучили реакции некоторых модельных соединений лигнина, и результаты были суммированы на рис.
Примечательно, что разрыв связей C sp2 –C sp3 в звеньях G и S затруднен из-за стерических и электронных эффектов, что сдерживало образование соответственно 2-метоксифенола и 2,6-диметоксифенола и обеспечивало высокую селективность по фенолу ( 13 ). Нами были также проведены трансформации лигнинов, выделенных из других пород древесины, в том числе из сосны, ивы, кипариса и травянистого растения Phyllostachys pubescens (рис.С8). Выходы фенола составили соответственно 9,0, 6,3, 4,5 и 10,1% (в пересчете на лигнин) при высокой селективности. Чтобы получить чистый фенол, мы провели масштабный эксперимент по трансформации лигнина тополя (рис. S9), в результате которого из 50,0 г лигнина было получено 4,1 г чистого фенола. Выход фенола был несколько снижен по сравнению с результатом газовой хроматографии (ГХ), в основном из-за потерь в процессе разделения. Чтобы дополнительно доказать эффективность катализатора, мы изучили реакции некоторых модельных соединений лигнина, и результаты были суммированы на рис. 2Б. Облигация C SP2 -C SP2 -C SP3 -C SP3 в H-производных ( 1A , 2A , и 3A ), G-производные ( 4A , 5A и 6A ), а S производные ( 7a , 8a и 9a ) модельные соединения могут быть эффективно расщеплены, что подтверждает практичность этой стратегии при разрыве связи C sp2 –C sp3 . Кроме того, продукты из модельных соединений димерного лигнина ( 3a , 6a и 9a ) также показали, что фенольный гидроксил (–OH) может быть образован гидролизом алифатической связи C β –O, что согласуется с выводом о том, что твердый кислотный катализатор может эффективно способствовать гидролизу ( 35 ).Результаты также показывают, что протокол производства фенола из лигнина, предложенный в этой работе (рис. 1C), осуществим.
2Б. Облигация C SP2 -C SP2 -C SP3 -C SP3 в H-производных ( 1A , 2A , и 3A ), G-производные ( 4A , 5A и 6A ), а S производные ( 7a , 8a и 9a ) модельные соединения могут быть эффективно расщеплены, что подтверждает практичность этой стратегии при разрыве связи C sp2 –C sp3 . Кроме того, продукты из модельных соединений димерного лигнина ( 3a , 6a и 9a ) также показали, что фенольный гидроксил (–OH) может быть образован гидролизом алифатической связи C β –O, что согласуется с выводом о том, что твердый кислотный катализатор может эффективно способствовать гидролизу ( 35 ).Результаты также показывают, что протокол производства фенола из лигнина, предложенный в этой работе (рис. 1C), осуществим. Рис. 2 Деконструкция лигнина и модельных соединений лигнина.
( A ) Разложение лигнина тополя. Условия реакции следующие: лигнин (0,4 г), HY 30 (0,4 г), H 2 O (5,0 мл), 200°С, 3 часа, 0,5 МПа Ar, 800 об/мин. ( B ) Деконструкция репрезентативных модельных соединений лигнина. Условия реакции следующие: субстрат (1 ммоль), цеолит HY 30 (0.3 г), H 2 O (4,0 мл), 180°C, 1 час, 0,5 МПа Ar, 800 об/мин. Все результаты реакции в (A) и (B) являются средними значениями трех экспериментов, проведенных параллельно.
Условия реакции следующие: лигнин (0,4 г), HY 30 (0,4 г), H 2 O (5,0 мл), 200°С, 3 часа, 0,5 МПа Ar, 800 об/мин. ( B ) Деконструкция репрезентативных модельных соединений лигнина. Условия реакции следующие: субстрат (1 ммоль), цеолит HY 30 (0.3 г), H 2 O (4,0 мл), 180°C, 1 час, 0,5 МПа Ar, 800 об/мин. Все результаты реакции в (A) и (B) являются средними значениями трех экспериментов, проведенных параллельно.
 Как показано на рис. 3, различные модели замещения алкила вместе с гидроксильной группой (от 10a до 15a ) в положении C α [C sp2 –C sp3 (OH)R], также переносились, и соответствующие продукты расщепления (от 10b до 15b ) были получены с выходами от умеренных до хороших (от 70 до 95%). Эффективность разрушения была почти равна эффективности 1a , когда водород в положении C α заменен на CH 3 ( 16a ), с 95.Выход анизола 0% ( 16b ). Производное 1a без заместителя в бензольном кольце ( 17a ) также может давать продукт деструкции 17b с выходом 92,7%. Когда гидроксильная группа в 1a изомеризуется в алифатическом положении C β ( 18a ), связь C sp2 –C sp3 также может разрушаться, но реакционная способность ниже с выходом 31,1. % ( 18b ). Кроме того, в нескольких структурно и электронно различных C α -замещенных (C sp2 –C sp3 X) связях, включая галогенсодержащие ( 19a и 20a ), сложноэфирные ( 21a и ) и алкенильных ( 23a и 24a ) групп, связь C sp2 –C sp3 также могла быть расщеплена, но эффективность была относительно ниже ( 19b – 2)7, чем с 6b 2 гидроксильная группа ( 1a ).
Как показано на рис. 3, различные модели замещения алкила вместе с гидроксильной группой (от 10a до 15a ) в положении C α [C sp2 –C sp3 (OH)R], также переносились, и соответствующие продукты расщепления (от 10b до 15b ) были получены с выходами от умеренных до хороших (от 70 до 95%). Эффективность разрушения была почти равна эффективности 1a , когда водород в положении C α заменен на CH 3 ( 16a ), с 95.Выход анизола 0% ( 16b ). Производное 1a без заместителя в бензольном кольце ( 17a ) также может давать продукт деструкции 17b с выходом 92,7%. Когда гидроксильная группа в 1a изомеризуется в алифатическом положении C β ( 18a ), связь C sp2 –C sp3 также может разрушаться, но реакционная способность ниже с выходом 31,1. % ( 18b ). Кроме того, в нескольких структурно и электронно различных C α -замещенных (C sp2 –C sp3 X) связях, включая галогенсодержащие ( 19a и 20a ), сложноэфирные ( 21a и ) и алкенильных ( 23a и 24a ) групп, связь C sp2 –C sp3 также могла быть расщеплена, но эффективность была относительно ниже ( 19b – 2)7, чем с 6b 2 гидроксильная группа ( 1a ).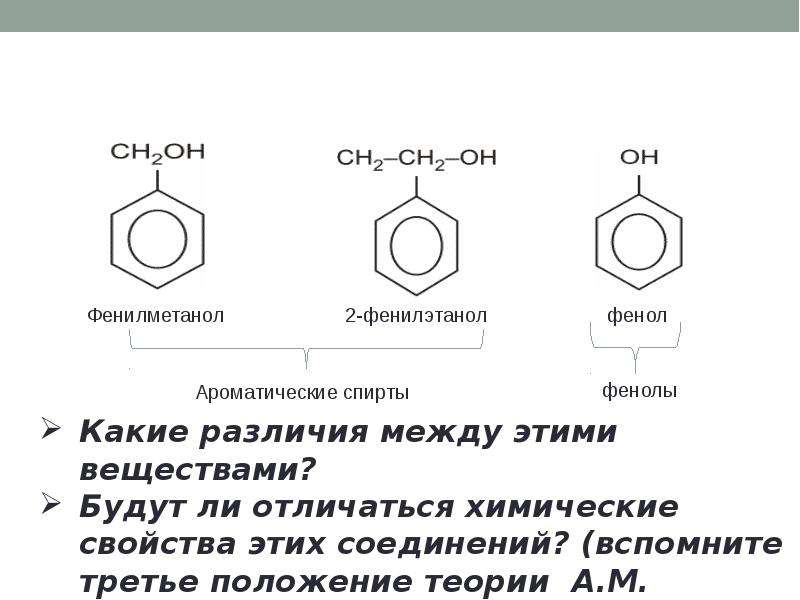 Для сравнения, p -гидроксибензойная кислота ( 25a ) подвергалась декарбоксилированию с образованием фенола ( 25b ) с выходом 90,6%, демонстрируя, что разрушение связи C sp2 –C sp2 с карбонильной средой при положение C α [C sp2 – C sp2 (=O)OR] может быть эффективно доступно с помощью этой стратегии. Учитывая вышеупомянутый разрыв связи C sp2 –C sp3 с различными средами C α , достоинство этой стратегии проявляется в деконструкции связей C sp2 –C sp3 с гидроксильной группой при положение С α , благоприятное для трансформации связи С sp2 – С sp3 в структуре лигнина.
Для сравнения, p -гидроксибензойная кислота ( 25a ) подвергалась декарбоксилированию с образованием фенола ( 25b ) с выходом 90,6%, демонстрируя, что разрушение связи C sp2 –C sp2 с карбонильной средой при положение C α [C sp2 – C sp2 (=O)OR] может быть эффективно доступно с помощью этой стратегии. Учитывая вышеупомянутый разрыв связи C sp2 –C sp3 с различными средами C α , достоинство этой стратегии проявляется в деконструкции связей C sp2 –C sp3 с гидроксильной группой при положение С α , благоприятное для трансформации связи С sp2 – С sp3 в структуре лигнина.Рис. 3. Прямая деконструкция связи C sp2 –C sp3 с различными связующими средами в алифатическом положении C α .
Результаты реакции представляют собой средние значения трех экспериментов, проведенных параллельно.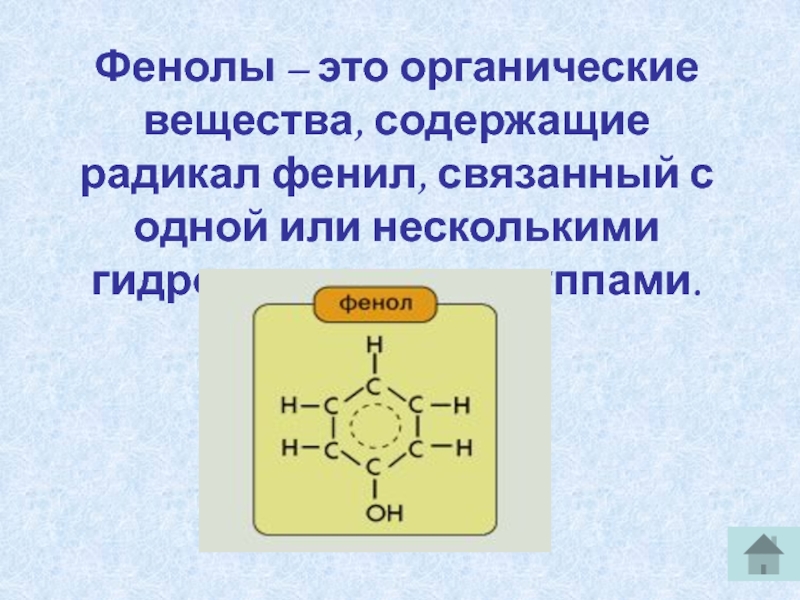 Условия реакции следующие: субстрат (1 ммоль), HY 30 (0,3 г), H 2 O (4,0 мл), 180°С, 1 час, 0,5 МПа Ar, 800 об/мин. *Bzl, бензильная группа; †Ac, ацетильная группа; ‡Bz, бензоильная группа.
Условия реакции следующие: субстрат (1 ммоль), HY 30 (0,3 г), H 2 O (4,0 мл), 180°С, 1 час, 0,5 МПа Ar, 800 об/мин. *Bzl, бензильная группа; †Ac, ацетильная группа; ‡Bz, бензоильная группа.
 Расчетный профиль реакции показан на рис. 4. Гидроксильная группа C α первоначально протонируется по кислотному центру Бренстеда HY 30 , что приводит к образованию иона оксония (структура I), который затем превращается в карбониевую группу. ион (структура II) с отщеплением H 2 O. Затем термодинамически γ-метил в боковой цепи смещается в алифатическое положение C α функцией цеолита, превращаясь в химически адсорбированную структуру III с миграционный барьер 1.18 эВ (TS-1), и последующее отщепление протона цеолитом дает структуру IV с более низким барьером 0,88 эВ (TS-2). Затем структура IV подвергается второму протонированию в положении C α с образованием иона карбония V (структура V), и это будет преследоваться ловушкой H 2 O, что дает геометрию третичного спирта (структура VI) и одновременно осуществляет еще одно протонное омоложение кислотного центра Бренстеда. После этого эндотермический поворот бензольного кольца третичного спирта (структура VI) к кислотному центру Бренстеда дает соответствующую геометрию (структура VII), позволяющую протонировать положение C sp2 (структура VIII) с эффективным барьером 0.
Расчетный профиль реакции показан на рис. 4. Гидроксильная группа C α первоначально протонируется по кислотному центру Бренстеда HY 30 , что приводит к образованию иона оксония (структура I), который затем превращается в карбониевую группу. ион (структура II) с отщеплением H 2 O. Затем термодинамически γ-метил в боковой цепи смещается в алифатическое положение C α функцией цеолита, превращаясь в химически адсорбированную структуру III с миграционный барьер 1.18 эВ (TS-1), и последующее отщепление протона цеолитом дает структуру IV с более низким барьером 0,88 эВ (TS-2). Затем структура IV подвергается второму протонированию в положении C α с образованием иона карбония V (структура V), и это будет преследоваться ловушкой H 2 O, что дает геометрию третичного спирта (структура VI) и одновременно осуществляет еще одно протонное омоложение кислотного центра Бренстеда. После этого эндотермический поворот бензольного кольца третичного спирта (структура VI) к кислотному центру Бренстеда дает соответствующую геометрию (структура VII), позволяющую протонировать положение C sp2 (структура VIII) с эффективным барьером 0. 57 эВ (ТС-3). Образовавшийся карбониевый ион VIII подвергается желаемому разрыву β-связи C sp2 – C sp3 с экзотермическим образованием фенола и ацетона ( 21 ).
57 эВ (ТС-3). Образовавшийся карбониевый ион VIII подвергается желаемому разрыву β-связи C sp2 – C sp3 с экзотермическим образованием фенола и ацетона ( 21 ). Рис. 4 Механистические исследования.
Энергетический профиль прямого разрушения связи C sp2 –C sp3 на катализаторе HY 30 . Промежуточные продукты реакции (от I до VIII) и переходное состояние (TS, от 1 до 3). Zeo, HY 30 цеолит.
В поддержку этого механизма было проведено испытание мечения изотопов 1a в H 2 18 O (рис.S10), а молекулярная масса пропанона составляла 60, а именно, был получен пропан-2-( 18 O) 1 , подтверждая, что стратегия деконструкции первоначально протекает с легким процессом дегидроксилирования, но затем снова гидроксилируется в алифатической C α положение с использованием молекулы воды. В ходе исследований по оптимизации (рис. S4) мы также обнаружили, что конверсия 1a увеличивалась быстрее, чем выход фенола, что указывает на то, что γ-метильный сдвиг является этапом, определяющим скорость деконструкции C sp2. –C sp3 , что согласуется с исследованиями механизма DFT.
–C sp3 , что согласуется с исследованиями механизма DFT.
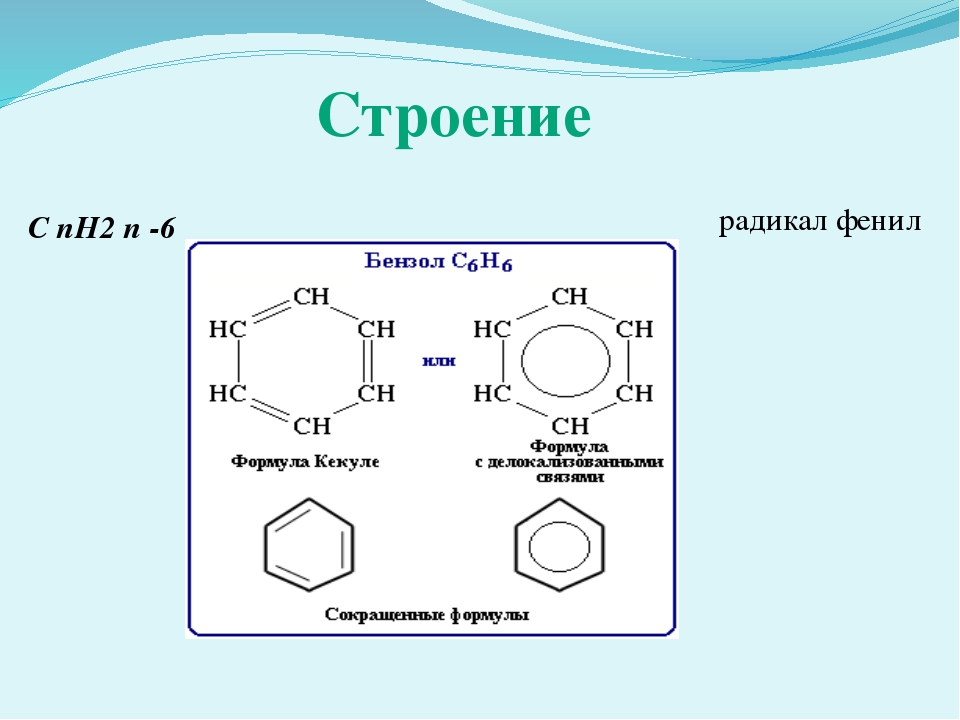 S12 и таблицу S5). В частности, сигналы 13 C, связанные с исходными атомами C β1 и C β2 при 24,5 и 21,0 м.д., сильно коррелируют с сигналами 1 H при 1,5 и 1,9 м.д., которые должны быть связаны с (–C β1 H 2 –) и (–C β2 H 3 ) в интермедиатах II и III соответственно. Наблюдаемые сигналы 13 C и 1 H при 12,7 и 0,7 м.д. относятся к фрагменту (–C γ H 3 ) в интермедиате II.В соответствии с анализом ЯМР 13 C (рис. 5C), 2D 13 C{ 1 H} HETCOR ЯМР дает хорошо разрешенный коррелированный сигнал при 31,2 м.д. в измерении 13 C и при 2,0 м.д. в размерности 1 H, что опять же отражает перенесенный атом C γ , а именно C β3 из фрагмента (–C β3 H 2 -Zeo) в интермедиате III. В частности, считается, что химическая связь с атомом кислорода в каркасном звене [≡Al–O–Si≡] обеспечивает индуктивный эффект для атома C β3 , который сдвигает его резонанс в слабое поле по сравнению с исходным C .
S12 и таблицу S5). В частности, сигналы 13 C, связанные с исходными атомами C β1 и C β2 при 24,5 и 21,0 м.д., сильно коррелируют с сигналами 1 H при 1,5 и 1,9 м.д., которые должны быть связаны с (–C β1 H 2 –) и (–C β2 H 3 ) в интермедиатах II и III соответственно. Наблюдаемые сигналы 13 C и 1 H при 12,7 и 0,7 м.д. относятся к фрагменту (–C γ H 3 ) в интермедиате II.В соответствии с анализом ЯМР 13 C (рис. 5C), 2D 13 C{ 1 H} HETCOR ЯМР дает хорошо разрешенный коррелированный сигнал при 31,2 м.д. в измерении 13 C и при 2,0 м.д. в размерности 1 H, что опять же отражает перенесенный атом C γ , а именно C β3 из фрагмента (–C β3 H 2 -Zeo) в интермедиате III. В частности, считается, что химическая связь с атомом кислорода в каркасном звене [≡Al–O–Si≡] обеспечивает индуктивный эффект для атома C β3 , который сдвигает его резонанс в слабое поле по сравнению с исходным C .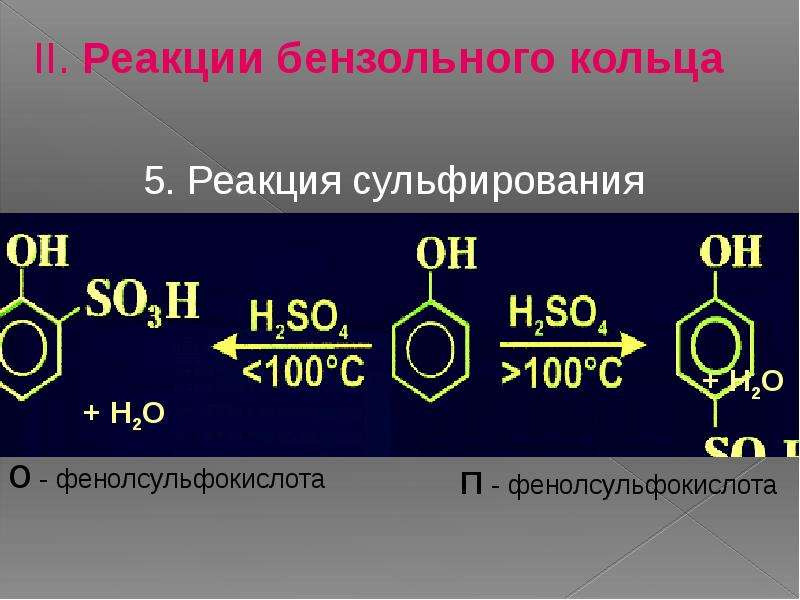 атомы β1 и C β2 , поддерживающие катализируемый цеолитом механизм γ-метильного сдвига ( 39 , 40 ).Такое промежуточное звено [≡Al–O(C β3 )–Si≡], относящееся к новому атому C β3 и атому кислорода цеолита, также было подтверждено анализами 27 Al ЯМР и инфракрасной спектроскопии с преобразованием Фурье (FTIR). (Рис. 5, D и E, и рис. S14). Как портной кислотности каркаса, тетраэдрически координированный Al [Al(IV)] в каркасе исходного и прокипяченного цеолитов всегда появляется одиночный резонанс при ок. 59,7 частей на миллион (рис. 5D и рис. S14A) ( 41 ). Сигнал плечевого резонанса на уровне ок.В рабочем цеолите идентифицировано 51,8 ppm, что можно отнести к атому Al(IV) в искаженном окружении [≡Al–O(C β3 )–Si≡] (рис. 5Д). Компенсация заряда атома C β3 ослабляет индуктивное влияние атома O от соседнего атома Al(IV), что уменьшает валентный угол Al–O–Si и удлиняет связи Al–O и Si–O, сдвигая резонанс в сильное поле от атома Al(IV) в регулярных [≡Al–O–Si≡] окружениях ( 42 – 44 ).
атомы β1 и C β2 , поддерживающие катализируемый цеолитом механизм γ-метильного сдвига ( 39 , 40 ).Такое промежуточное звено [≡Al–O(C β3 )–Si≡], относящееся к новому атому C β3 и атому кислорода цеолита, также было подтверждено анализами 27 Al ЯМР и инфракрасной спектроскопии с преобразованием Фурье (FTIR). (Рис. 5, D и E, и рис. S14). Как портной кислотности каркаса, тетраэдрически координированный Al [Al(IV)] в каркасе исходного и прокипяченного цеолитов всегда появляется одиночный резонанс при ок. 59,7 частей на миллион (рис. 5D и рис. S14A) ( 41 ). Сигнал плечевого резонанса на уровне ок.В рабочем цеолите идентифицировано 51,8 ppm, что можно отнести к атому Al(IV) в искаженном окружении [≡Al–O(C β3 )–Si≡] (рис. 5Д). Компенсация заряда атома C β3 ослабляет индуктивное влияние атома O от соседнего атома Al(IV), что уменьшает валентный угол Al–O–Si и удлиняет связи Al–O и Si–O, сдвигая резонанс в сильное поле от атома Al(IV) в регулярных [≡Al–O–Si≡] окружениях ( 42 – 44 ).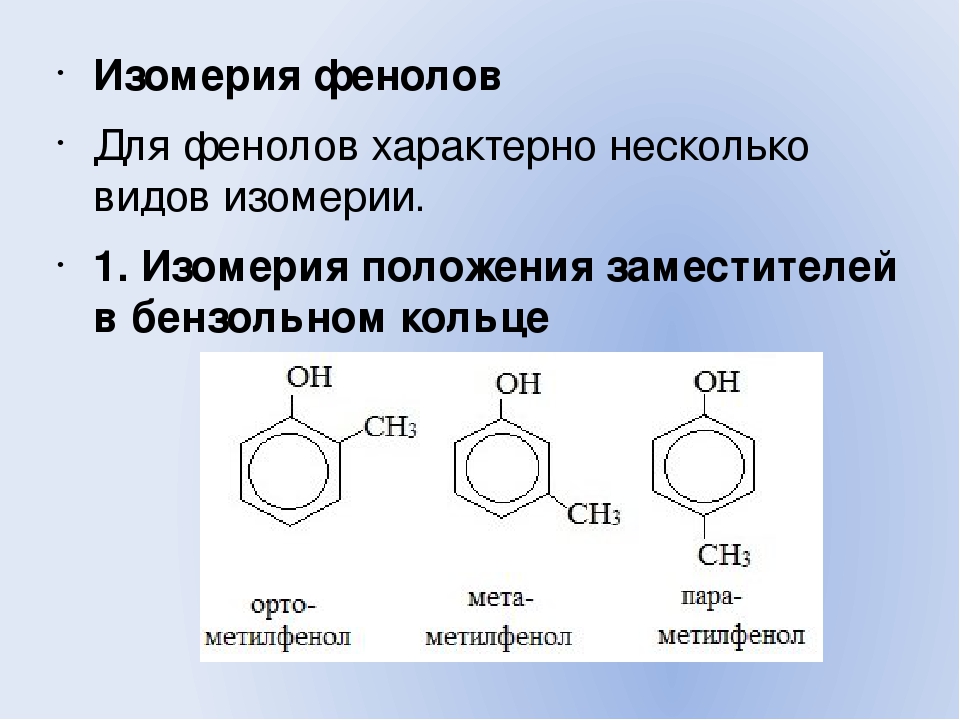 В спектрах FTIR (рис.S14B), интенсивность кислой группы Бренстеда [≡Al–OH–Si≡] при 3630 см –1 явно уменьшилась для цеолита в реакции ( 45 ), и, соответственно, заметное красное смещение в инфракрасной частоте относительно асимметричного валентного колебания ячейки решетки [≡Al–O–Si≡], которое также должно быть связано с удлинением связей Al–O и Si–O, обусловленным взаимодействием между атомами кислорода цеолита в звеньев каркаса [≡Al–O–Si≡] и атомов C β3 ( 46 ), что хорошо соответствует анализу ЯМР 27 Al.Приведенные выше определения прекрасно согласуются с исследованиями механизма DFT (рис. 4), в которых γ-метильный сдвиг предлагается в качестве решающего шага для формирования геометрии третичного спирта (структура VI), которая приносит пользу конечному C sp2 – C sp3 разрыв связи β (TS-3; 0,57 эВ). Для сравнения, при замене γ-метила на атом H в структурной формуле разрыв связи C sp2 –C sp3 в 1-(4-метоксифенил)этаноле (рис.
В спектрах FTIR (рис.S14B), интенсивность кислой группы Бренстеда [≡Al–OH–Si≡] при 3630 см –1 явно уменьшилась для цеолита в реакции ( 45 ), и, соответственно, заметное красное смещение в инфракрасной частоте относительно асимметричного валентного колебания ячейки решетки [≡Al–O–Si≡], которое также должно быть связано с удлинением связей Al–O и Si–O, обусловленным взаимодействием между атомами кислорода цеолита в звеньев каркаса [≡Al–O–Si≡] и атомов C β3 ( 46 ), что хорошо соответствует анализу ЯМР 27 Al.Приведенные выше определения прекрасно согласуются с исследованиями механизма DFT (рис. 4), в которых γ-метильный сдвиг предлагается в качестве решающего шага для формирования геометрии третичного спирта (структура VI), которая приносит пользу конечному C sp2 – C sp3 разрыв связи β (TS-3; 0,57 эВ). Для сравнения, при замене γ-метила на атом H в структурной формуле разрыв связи C sp2 –C sp3 в 1-(4-метоксифенил)этаноле (рис.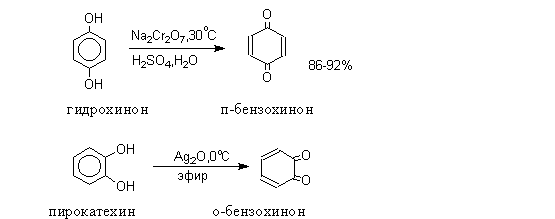 3, 10a ) также может происходить через протонирование положения C sp2 в геометрии первичного спирта, но с более высоким энергетическим барьером 0.72 эВ (рис. S14) кинетически снижала эффективность разрыва связи C sp2 –C sp3 , что хорошо согласуется с экспериментальным результатом.
3, 10a ) также может происходить через протонирование положения C sp2 в геометрии первичного спирта, но с более высоким энергетическим барьером 0.72 эВ (рис. S14) кинетически снижала эффективность разрыва связи C sp2 –C sp3 , что хорошо согласуется с экспериментальным результатом. Рис. 5 Структурный анализ эволюции связи С–С в реакции 1а.
( A ) Заполняющая пространство модель структуры III на рис. 3. ( B ) Метрики основных связей структуры III. ( C ) 13 C Спектр ЯМР промежуточных продуктов реакции 1a ; черный сплошной квадрат (■) представляет наблюдаемые интенсивности; фиолетовые круглые метки (○) — общая кривая подгонки; темно-желтый правильный треугольник вверх (△), розовый правильный треугольник вниз (▽), голубой полый левый треугольник (◁), зеленый ромб (◊) и оливковый полый левый треугольник (▷) представляют аппроксимирующие кривые C α , C β1 , С β2 , С β3 и С γ соответственно. ( D ) 27 Al Спектр ЯМР исходного цеолита HY 30 ; черный сплошной квадрат (■) представляет наблюдаемые интенсивности; синий перевернутый треугольник (▽) — кривая подгонки каркаса Al(IV). ( E ) 27 Al Спектр ЯМР цеолита HY 30 в реакции; черный сплошной квадрат (■) представляет наблюдаемые интенсивности; фиолетовые круглые метки (○) — общая кривая подгонки; синий перевернутый треугольник (▽) и зелено-желтый правильный треугольник (△) представляют собой аппроксимирующие кривые регулярного каркаса Al(IV) и Al(IV) связывания с C β2 соответственно.Условия реакции следующие: 1a (1 ммоль), HY 30 (0,3 г), H 2 O (4,0 мл), 180°C, 10 мин, 0,5 МПа Ar, 800 об/мин. анализы ЯМР 13 C (рис. 5C, фиг. S11A и S12A, и таблицы S4 и S5), резонансный сигнал для алифатического атома C α в 1a при 73,9 м.д. исчез и сместился до 36,7 м.д., которые подтвердили, что гидроксильная группа в алифатическом положении C α может быть протонирована и быстро удалена в реакции, что согласуется с исследованиями DFT.
( D ) 27 Al Спектр ЯМР исходного цеолита HY 30 ; черный сплошной квадрат (■) представляет наблюдаемые интенсивности; синий перевернутый треугольник (▽) — кривая подгонки каркаса Al(IV). ( E ) 27 Al Спектр ЯМР цеолита HY 30 в реакции; черный сплошной квадрат (■) представляет наблюдаемые интенсивности; фиолетовые круглые метки (○) — общая кривая подгонки; синий перевернутый треугольник (▽) и зелено-желтый правильный треугольник (△) представляют собой аппроксимирующие кривые регулярного каркаса Al(IV) и Al(IV) связывания с C β2 соответственно.Условия реакции следующие: 1a (1 ммоль), HY 30 (0,3 г), H 2 O (4,0 мл), 180°C, 10 мин, 0,5 МПа Ar, 800 об/мин. анализы ЯМР 13 C (рис. 5C, фиг. S11A и S12A, и таблицы S4 и S5), резонансный сигнал для алифатического атома C α в 1a при 73,9 м.д. исчез и сместился до 36,7 м.д., которые подтвердили, что гидроксильная группа в алифатическом положении C α может быть протонирована и быстро удалена в реакции, что согласуется с исследованиями DFT. Можно сделать вывод, что лабильная химия гидроксильной группы в алифатическом положении C α [C sp2 – C sp3 (OH)] является триггером для эффективного расщепления C sp2 – C . sp3 в таких мягких условиях, которые способствовали одновременному разрушению связей C sp2 –C sp3 и гидролизу алифатических связей C β –O и в конечном итоге реализовали прямое отщепление фенола от структуры лигнина (рис. .S6, А и Б). Напротив, в ходе обычных химических воронкообразных процессов (рис. 1А) расщепление связей C sp2 –C sp3 в алкилфенолах, полученных из лигнинового масла, без гидроксильной группы в положении C α всегда требовало значительно большей температура реакции ( 12 ). В строительных блоках, полученных из H (рис. S7 и S15), алифатические положения C α всегда имеют замещенные гидроксильные группы ( 33 , 47 ), что, следовательно, обеспечивает возможность прямой деконструкции кинетически инертного C –С связь в мягких условиях для превращения лигнина в фенол.
Можно сделать вывод, что лабильная химия гидроксильной группы в алифатическом положении C α [C sp2 – C sp3 (OH)] является триггером для эффективного расщепления C sp2 – C . sp3 в таких мягких условиях, которые способствовали одновременному разрушению связей C sp2 –C sp3 и гидролизу алифатических связей C β –O и в конечном итоге реализовали прямое отщепление фенола от структуры лигнина (рис. .S6, А и Б). Напротив, в ходе обычных химических воронкообразных процессов (рис. 1А) расщепление связей C sp2 –C sp3 в алкилфенолах, полученных из лигнинового масла, без гидроксильной группы в положении C α всегда требовало значительно большей температура реакции ( 12 ). В строительных блоках, полученных из H (рис. S7 и S15), алифатические положения C α всегда имеют замещенные гидроксильные группы ( 33 , 47 ), что, следовательно, обеспечивает возможность прямой деконструкции кинетически инертного C –С связь в мягких условиях для превращения лигнина в фенол.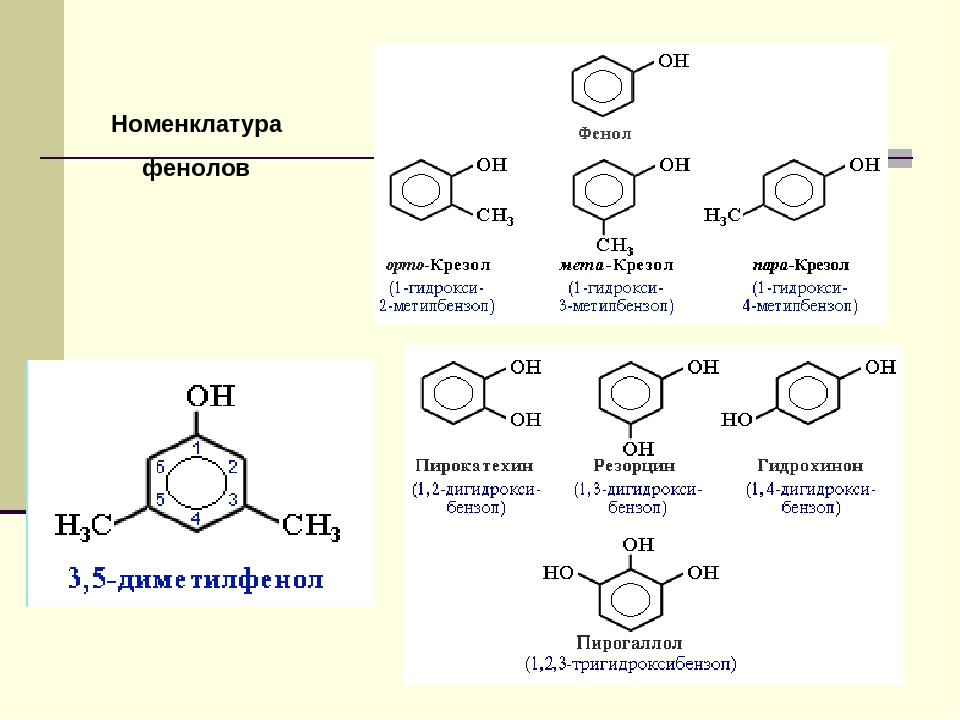 Кроме того, на основе количественного анализа 13 C (рис. S15B) количество H-производных строительных блоков на 100 ароматических единиц в экстрагированном лигнине тополя составило 3,0 в форме p -гидроксибензоата (PB ) единица измерения. После реакции сигналы ЯМР 13 C для всех H-производных строительных блоков, включая звенья PB, исчезли в остатке лигнина (рис. S15C), что подтверждает образование фенола.
Кроме того, на основе количественного анализа 13 C (рис. S15B) количество H-производных строительных блоков на 100 ароматических единиц в экстрагированном лигнине тополя составило 3,0 в форме p -гидроксибензоата (PB ) единица измерения. После реакции сигналы ЯМР 13 C для всех H-производных строительных блоков, включая звенья PB, исчезли в остатке лигнина (рис. S15C), что подтверждает образование фенола. МАТЕРИАЛЫ И МЕТОДЫ
Материалы
4′-Метоксипропиофенон (99%), 4′-метоксиацетофенон (99%), 3′,4′-диметоксиацетофенон (98+%), 3′,4′,5′-триметоксиацетофенон (99%), 4′-метоксибутирофенон (97%), 4-метоксифенилацетон (97+%), 4-метоксибензальдегид (98%), 4-гидроксибензальдегид (98%), 3,4-диметоксибензальдегид (99%), 4 -гидрокси-3-метоксибензальдегид (99%), 3,4,5-триметоксибензальдегид (98%), 4′-гидрокси-3′,5′-диметоксибензальдегид (97%), 2-метоксифенол (98+%), бромэтан (98%), 1-бромгексан (99%), 1-бромдекан (98%), 2-фенилэтилбромид (98%), 1-бром-3-фенилпропан (98%), (±)-1-фенил- 1-пропанол (98+%), магниевая стружка (99+%), 4- n -пропилфенол (98%), бензоилхлорид (99+%), тионилхлорид (99+%), трибромид фосфора (99% ), триэтиламин (99+%), морденит 10 цеолит аммония, оксид алюминия (99. 5%), диоксид кремния (99,5%), хлорид рутения(III) (безводный; Ru 47,7%), гексагидрат нитрата никеля(II) (98%), октагидрат оксида дихлорида циркония (98%), оксид ниобия(V) ( 99,5%), ацетилацетонат хрома(III) (97%), дигидрат щавелевой кислоты (98%) и 4-гидроксибензойную кислоту (99%) были приобретены у Alfa Aesar. HY 3 (Si/Al = 3), HY 15 , HY 30 и HY 40 были синтезированы Alfa Aesar. Раствор гидроксида аммония (от 28 до 30%), раствор хлорида метилмагния (3.0 М в тетрагидрофуране) и диметилсульфат (99%) были приобретены у Beijing InnoChem Science and Technology Co. Ltd. Гидрат метавольфрамата аммония (99,9%) был приобретен у Strem Chemicals. 1-(4-Метоксифенил)-1-пропен был приобретен у Ark Pharm Inc. 1-Метокси-4-(1-пропен-2-ил)бензол был получен у Accela ChemBio Co. Ltd. ZSM-5 9 , ZSM-5 30 , ZSM-5 63 , ZSM-5 235 , Mordenite 13 , Beta 13 , Beta 30 , Beta 50 , и MCM-41 Zeolites были приобретены из Нанкая Университет Катализатор Ко.
5%), диоксид кремния (99,5%), хлорид рутения(III) (безводный; Ru 47,7%), гексагидрат нитрата никеля(II) (98%), октагидрат оксида дихлорида циркония (98%), оксид ниобия(V) ( 99,5%), ацетилацетонат хрома(III) (97%), дигидрат щавелевой кислоты (98%) и 4-гидроксибензойную кислоту (99%) были приобретены у Alfa Aesar. HY 3 (Si/Al = 3), HY 15 , HY 30 и HY 40 были синтезированы Alfa Aesar. Раствор гидроксида аммония (от 28 до 30%), раствор хлорида метилмагния (3.0 М в тетрагидрофуране) и диметилсульфат (99%) были приобретены у Beijing InnoChem Science and Technology Co. Ltd. Гидрат метавольфрамата аммония (99,9%) был приобретен у Strem Chemicals. 1-(4-Метоксифенил)-1-пропен был приобретен у Ark Pharm Inc. 1-Метокси-4-(1-пропен-2-ил)бензол был получен у Accela ChemBio Co. Ltd. ZSM-5 9 , ZSM-5 30 , ZSM-5 63 , ZSM-5 235 , Mordenite 13 , Beta 13 , Beta 30 , Beta 50 , и MCM-41 Zeolites были приобретены из Нанкая Университет Катализатор Ко. ООО Кремневольфрамовая кислота, фосфорно-вольфрамовая кислота, фосфоромолибденовая кислота, соляная кислота (37%), серная кислота (98%), фосфорная кислота (85%), уксусный ангидрид (98,5%), боргидрид натрия (98%), бром (99,5% ), хлорид алюминия (99%) и раствор формальдегида (37%) были предоставлены Sinopharm Chemical Reagent Co. Ltd. Хлорид натрия (99,5%), сульфат магния (99,5%), хлорид аммония (99,5%), гидрокарбонат натрия (99,8%) и карбонат калия (99%) были приобретены у Aladdin Chemical Reagent Company.Тополь, сосна, ива и кипарис были получены из провинции Ганьсу, Китай. P. pubescens был получен из провинции Цзянси, Китай. Тетрагидрофуран (99,5%), этилацетат (99,5%), этанол (99,5%), пиридин (99,5%), эфир (99,5%) и ацетон (99,5%) были приобретены у Concord Technologies (Tianjin) Co. Ltd. Метилсульфоксид -d6 [Д; 99,9%, 0,03% (об./об.) тетраметилсилан (ТМС)] был приобретен у Cambridge Isotope Laboratories Inc. , 10%; 99.99%), и O 2 /Ar (O 2 , 1%; 99,99%) были предоставлены компанией Beijing Analytic Instrument Company.
ООО Кремневольфрамовая кислота, фосфорно-вольфрамовая кислота, фосфоромолибденовая кислота, соляная кислота (37%), серная кислота (98%), фосфорная кислота (85%), уксусный ангидрид (98,5%), боргидрид натрия (98%), бром (99,5% ), хлорид алюминия (99%) и раствор формальдегида (37%) были предоставлены Sinopharm Chemical Reagent Co. Ltd. Хлорид натрия (99,5%), сульфат магния (99,5%), хлорид аммония (99,5%), гидрокарбонат натрия (99,8%) и карбонат калия (99%) были приобретены у Aladdin Chemical Reagent Company.Тополь, сосна, ива и кипарис были получены из провинции Ганьсу, Китай. P. pubescens был получен из провинции Цзянси, Китай. Тетрагидрофуран (99,5%), этилацетат (99,5%), этанол (99,5%), пиридин (99,5%), эфир (99,5%) и ацетон (99,5%) были приобретены у Concord Technologies (Tianjin) Co. Ltd. Метилсульфоксид -d6 [Д; 99,9%, 0,03% (об./об.) тетраметилсилан (ТМС)] был приобретен у Cambridge Isotope Laboratories Inc. , 10%; 99.99%), и O 2 /Ar (O 2 , 1%; 99,99%) были предоставлены компанией Beijing Analytic Instrument Company.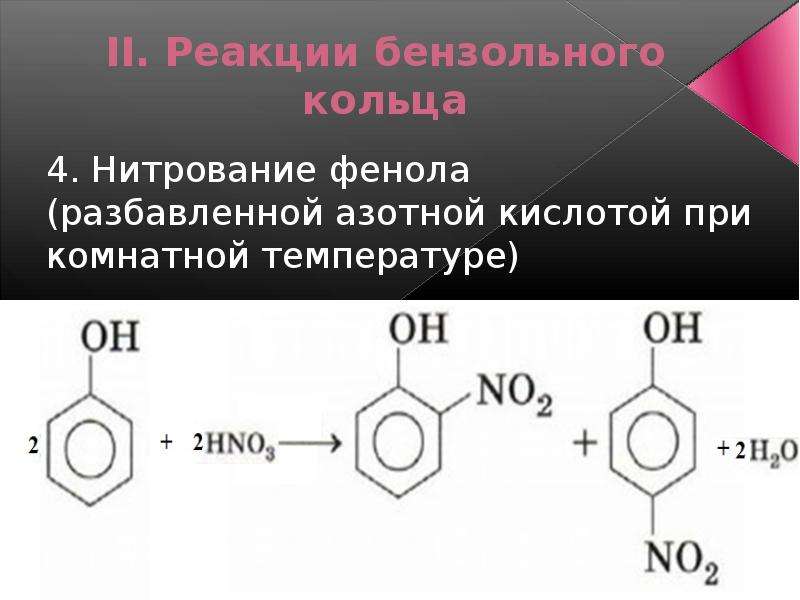 Деионизированная вода была предоставлена Институтом химии Китайской академии наук. Все реагенты использовали в том виде, в каком они были получены, без дополнительной очистки.
Деионизированная вода была предоставлена Институтом химии Китайской академии наук. Все реагенты использовали в том виде, в каком они были получены, без дополнительной очистки.
Catalyst Price
Зеолитные катализаторы
, HY 3 , HY 15 , HY 30 , HY 40 , ZSM-5 9 , ZSM-5 30 , ZSM-5 63 , ZSM-5 , ZSM-5 235 , Mordenite 10 аммоний, Mordenite 13 , бета- 13 , бета- 30 , бета 50 , и MCM-41 были предварительно обработаны прокаливанием при 550 ° C для 4 часы.
RU / SIO 2 , Ni / SiO 2 , и W / SIO 2 Catalysts
Поддерживают RU / SIO 2 , NI / SIO 2 , A / W / SIO 2 Катализаторы готовили общепринятым методом влажной пропитки. В типичном препарате SiO 2 сушили при 120°C в течение ночи перед пропиткой.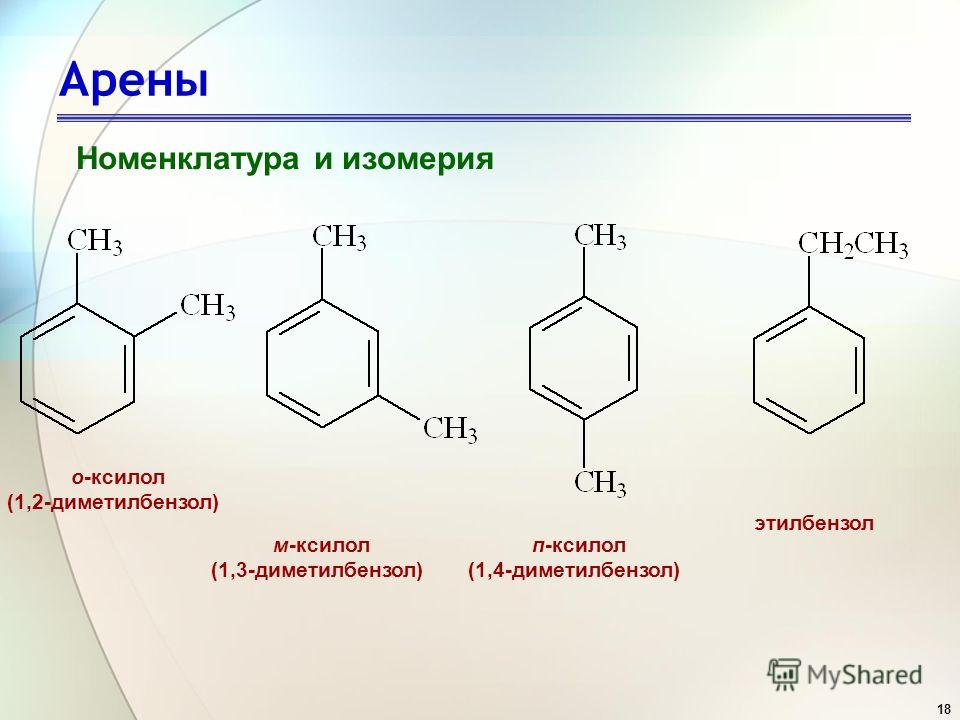 Хлорид рутения (III) (0,103 г), гексагидрат нитрата никеля (II) (0,248 г) и гидрат метавольфрамата аммония (0,067 г) растворяли в 10 мл деионизированной воды по отдельности.Опишем процедуры на примере Ru/SiO 2 . Раствор прекурсора последовательно по каплям добавляли к 30 мл деионизированной воды с 1 г аморфного SiO 2 при комнатной температуре. Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 200°С в течение 12 часов в сушильном шкафу. Свежеприготовленный образец восстанавливали в непрерывном потоке 10% H 2 /Ar при 350°C в течение 1 часа. После охлаждения до комнатной температуры в атмосфере аргона восстановленный катализатор выдерживали в атмосфере 1% O 2 /Ar в течение 1 часа для образования пассивирующего слоя, препятствующего объемному окислению перед выдержкой на воздухе.Катализаторы Ni/SiO 2 и W/SiO 2 на носителе готовили по тем же методикам, что и катализатор Ru/SiO 2 .
Хлорид рутения (III) (0,103 г), гексагидрат нитрата никеля (II) (0,248 г) и гидрат метавольфрамата аммония (0,067 г) растворяли в 10 мл деионизированной воды по отдельности.Опишем процедуры на примере Ru/SiO 2 . Раствор прекурсора последовательно по каплям добавляли к 30 мл деионизированной воды с 1 г аморфного SiO 2 при комнатной температуре. Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 200°С в течение 12 часов в сушильном шкафу. Свежеприготовленный образец восстанавливали в непрерывном потоке 10% H 2 /Ar при 350°C в течение 1 часа. После охлаждения до комнатной температуры в атмосфере аргона восстановленный катализатор выдерживали в атмосфере 1% O 2 /Ar в течение 1 часа для образования пассивирующего слоя, препятствующего объемному окислению перед выдержкой на воздухе.Катализаторы Ni/SiO 2 и W/SiO 2 на носителе готовили по тем же методикам, что и катализатор Ru/SiO 2 .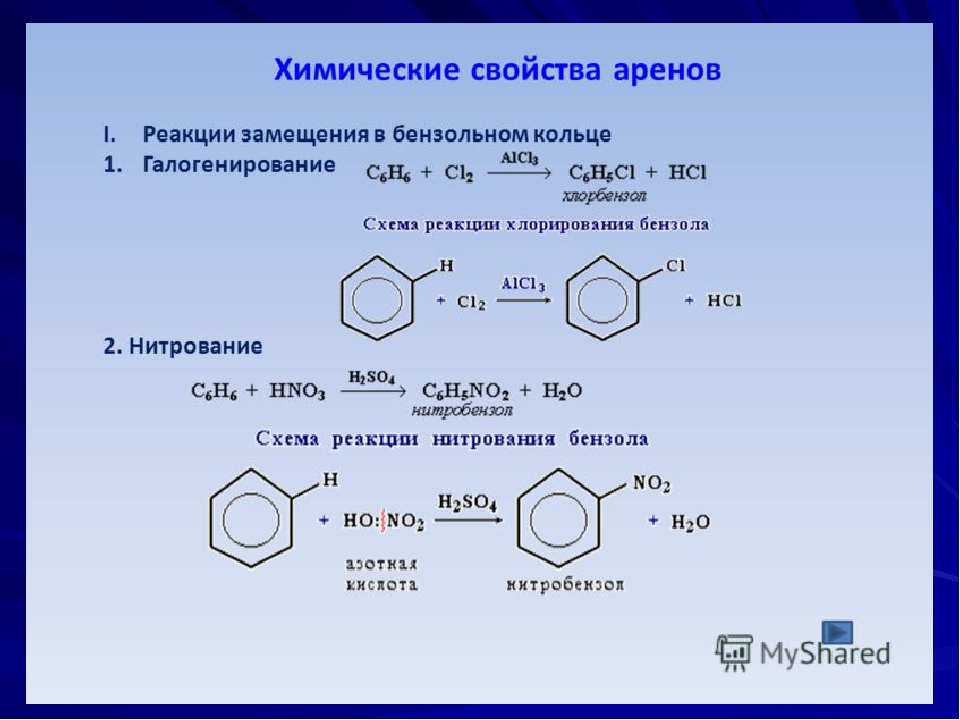 Приготовленные катализаторы были обозначены как катализаторы Ru/SiO 2 , Ni/SiO 2 и W/SiO 2 .
Приготовленные катализаторы были обозначены как катализаторы Ru/SiO 2 , Ni/SiO 2 и W/SiO 2 .
SO 2− 4 /ZrO 2 катализатор
Гидроксид циркония получали добавлением раствора гидроксида аммония в водный раствор октагидрата оксида дихлорида циркония.Осаждение происходило при значении рН до 10. Затем гель непрерывно перемешивали в течение 2 часов при комнатной температуре. После этого гель фильтровали, промывали деионизированной водой, а затем сушили при 120°С в течение 16 часов.
SO 2– 4 /ZrO 2 катализатор
В эксперименте последовательно по каплям добавляли 15 мл 0,05 М раствора серной кислоты -1 полученного гидроксида циркония при комнатной температуре.Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 120°С в течение 12 часов в сушильном шкафу. В последнюю очередь образец прокаливали в токе воздуха при 550°С в течение 2 часов. Приготовленный катализатор был обозначен как катализатор SO 2– 4 /ZrO 2 .
Приготовленный катализатор был обозначен как катализатор SO 2– 4 /ZrO 2 .
WO 3 /ZrO 2 катализатор
Гидрат метавольфрамата аммония (0,201 г) растворяли в 10 мл деионизированной воды. Затем раствор прекурсора последовательно по каплям добавляли в 30 мл деионизированной воды с 1 г полученного гидроксида циркония при комнатной температуре.Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 120°С в течение 12 часов в сушильном шкафу. В последнюю очередь образец прокаливали в токе воздуха при 600°С в течение 3 часов. Приготовленный катализатор был обозначен как катализатор WO 3 /ZrO 2 .
WO 3 /Al 2 O 3 катализатор
Гидрат метавольфрамата аммония (0,201 г) растворяли в 10 мл деионизированной воды. Затем раствор прекурсора последовательно по каплям добавляли к 30 мл деионизированной воды с 1 г оксида алюминия при 60°С.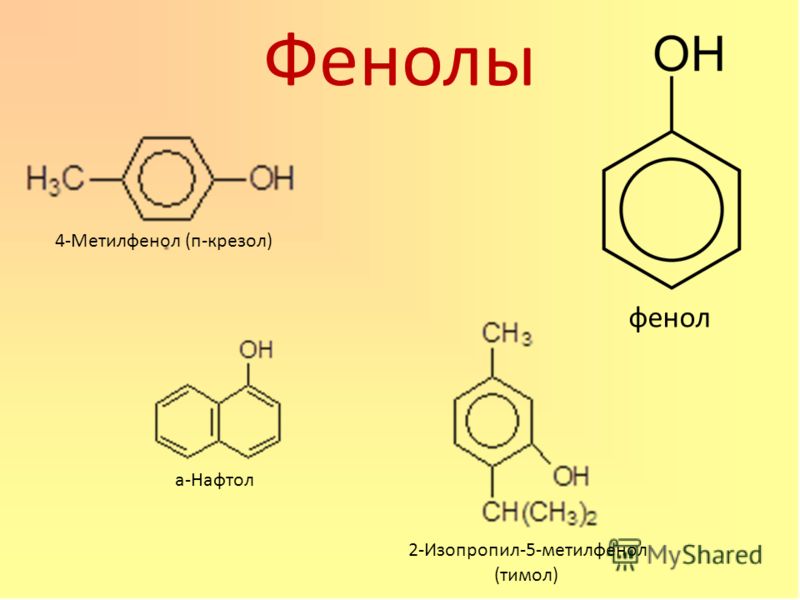 Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 120°С в течение 12 часов в сушильном шкафу. Наконец, образец прокалили в атмосфере воздуха при 600°С в течение 3 часов. Приготовленный катализатор был обозначен как катализатор WO 3 /Al 2 O 3 .
Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 120°С в течение 12 часов в сушильном шкафу. Наконец, образец прокалили в атмосфере воздуха при 600°С в течение 3 часов. Приготовленный катализатор был обозначен как катализатор WO 3 /Al 2 O 3 .
NB 2 NB 2 O 5 / AL 2 / AL 2 O 3 Catalyst 3 Catalyst
Catalited NB 2 O 5 / AL 2 O 3 Catalyst был подготовлен путем широко используемой влажной пропитки метод.Ниобиевую кислоту (0,3 г) растворяли в 10 мл водного раствора щавелевой кислоты. Затем раствор прекурсора последовательно по каплям добавляли в 30 мл деионизированной воды с 1 г оксида алюминия при комнатной температуре. Полученную смесь интенсивно перемешивали в течение 24 часов, упаривали в вакууме и сушили при 120°С в течение 12 часов в сушильном шкафу. В последнюю очередь образец прокаливали в токе воздуха при 500°С в течение 3 часов. Приготовленный катализатор был обозначен как катализатор Nb 2 O 5 /Al 2 O 3 .
В последнюю очередь образец прокаливали в токе воздуха при 500°С в течение 3 часов. Приготовленный катализатор был обозначен как катализатор Nb 2 O 5 /Al 2 O 3 .
Характеристика катализатора
Рентгенограммы (XRD) образцов были получены на рентгеновском дифрактометре Rigaku D/max 2500 с источником рентгеновского излучения Cu Kα и работали при 40 кВ и 200 мА. Диапазон 2θ сканировали от 15° до 40° со скоростью 2°/мин с шириной шага 0,02°.
Анализ термопрограммируемой десорбции аммиака (NH 3 -TPD) выполнен на анализаторе AutoChem 2950 HP. Все образцы нагревали до 400°С со скоростью 10°С мин -1 и выдерживали при 400°С в течение 1 часа.Адсорбцию аммиака на образцах проводили при комнатной температуре. После насыщения образцы продували током чистого азота при 100°С в течение 1 часа для удаления физически адсорбированного аммиака. Затем были получены профили NH 3 -TPD в диапазоне от 50° до 600°C при скорости нагрева 10°C мин -1 .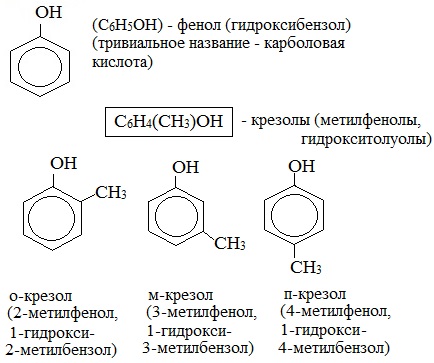
29 Si и 27 Al были получены на спектрометре Bruker Avance III 400 МГц с резонансным зондом 4 мм. Спектры MAS ЯМР 29 Si были записаны на резонансной частоте 79,30 МГц, с шириной импульса 1,5 мкс, временем задержки 2 с и скоростью вращения 8 кГц с 2500 сканированиями. Спектры MAS ЯМР 27 Al записывали на резонансной частоте 104.01 МГц, с шириной импульса 1 мкс, временем задержки 0,5 с и скоростью вращения 12 кГц с 8192 сканами.
Спектры MAS ЯМР 29 Si были записаны на резонансной частоте 79,30 МГц, с шириной импульса 1,5 мкс, временем задержки 2 с и скоростью вращения 8 кГц с 2500 сканированиями. Спектры MAS ЯМР 27 Al записывали на резонансной частоте 104.01 МГц, с шириной импульса 1 мкс, временем задержки 0,5 с и скоростью вращения 12 кГц с 8192 сканами.
N 2 Эксперименты по адсорбции-десорбции проводились с использованием Micromeritics TriStar II 3020 при температуре жидкого азота. Перед анализом образец дегазировали при 300°С в вакууме в течение 24 часов. Площади поверхности микропор/мезопор рассчитывали с использованием модели t -plot. Распределение пор по размерам в образце рассчитывали с использованием модели размеров пор Барретта-Джойнера-Халенды (BJH).Объемы микропор/мезопор рассчитывали с использованием моделей t и BJH соответственно.
Изображения, полученные с помощью сканирующей электронной микроскопии (СЭМ), были получены с использованием СЭМ S-4800, работающего при напряжении 10 кВ.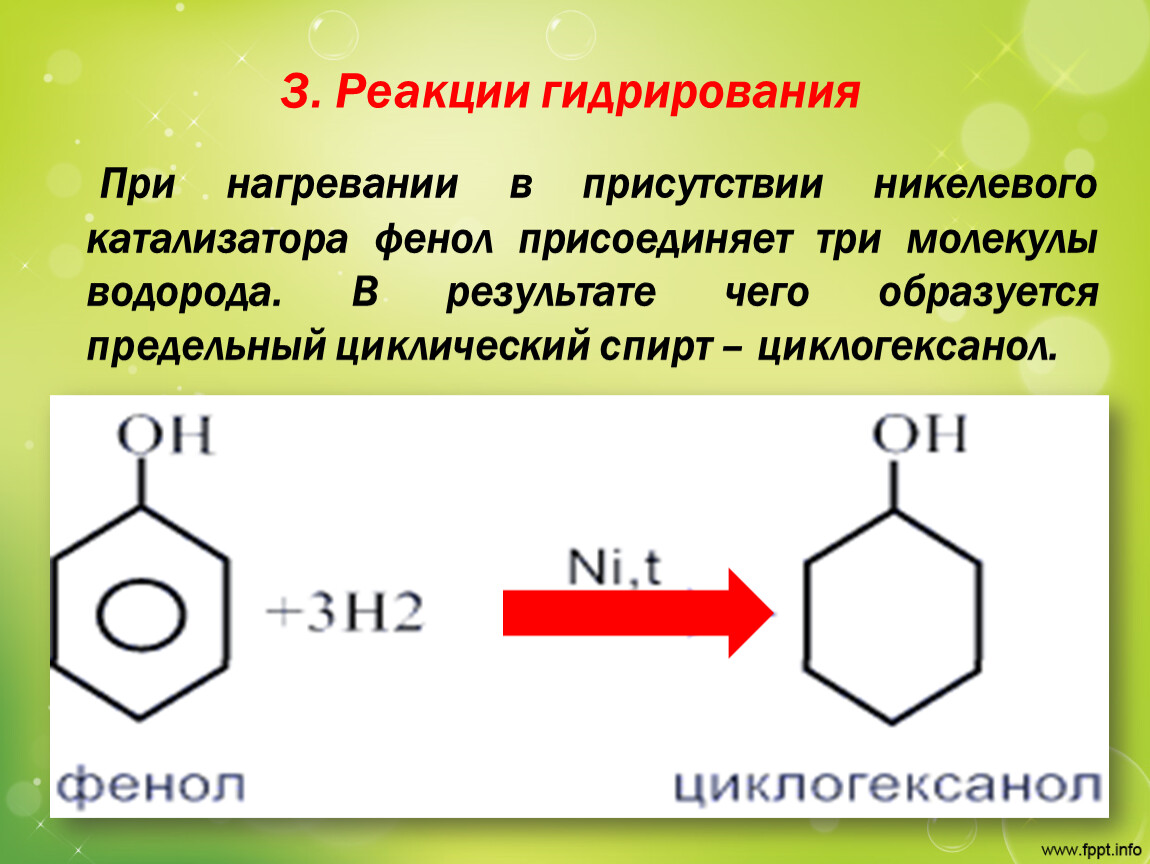 Изображения трансмиссионной электронной микроскопии (ПЭМ) были получены с использованием ПЭМ HT7700, работающего при 100 кВ. Содержание нанесенных металлов на катализаторы определяли с помощью индуктивно-связанной плазмы (Thermo Fisher Scientific, iCAP Q).
Изображения трансмиссионной электронной микроскопии (ПЭМ) были получены с использованием ПЭМ HT7700, работающего при 100 кВ. Содержание нанесенных металлов на катализаторы определяли с помощью индуктивно-связанной плазмы (Thermo Fisher Scientific, iCAP Q).
Экстракция и характеристика лигнина
Экстракцию лигнина проводили с использованием метода, аналогичного описанному в ( 49 ).Использовали 1-литровый автоклав (Weihai Xinyuan Chemical Machinery Co. Ltd.). В типичном эксперименте в автоклав загружали 70 г порошка тополя, 490 мл ацетона и 210 мл воды. Автоклав герметизировали и продували N 2 для удаления воздуха при комнатной температуре, а затем загружали N 2 под давлением 0,5 МПа. Затем автоклав нагревали до 160°С в течение 1 часа. После этого включали мешалку со скоростью перемешивания 600 об/мин и регистрировали время реакции.Через 1 час автоклав быстро охлаждали и выпускали газ. Жидкость собирали фильтрованием. Остаток на фильтре трижды промывали 200 мл ацетона/H 2 O (9:1), промывную жидкость собирали и объединяли с жидкостью фильтрата. После этого жидкость концентрировали под вакуумом при 30°С до помутнения жидкости и снова растворяли в 30 мл ацетона. Затем концентрированный раствор лигнина медленно вливали в быстро перемешиваемую 2000 мл воды и отфильтровывали осадок с помощью воронки с размером пор 3–4 мкм.Затем лигнин-сырец растворяли в 150 мл ацетона/H 2 O (9:1) и снова осаждали в воду тем же способом. Наконец, сырой лигнин растворяют в 200 мл раствора ацетон/метанол (9:1) и сушат над безводным MgSO 4 . Затем жидкость медленно вливали в быстро перемешиваемые 2000 мл диэтилового эфира и фильтровали с использованием воронки с размером пор от 3 до 4 мкм. Твердое вещество лигнина лиофилизировали в вакууме в течение 24 часов и в итоге получили 3,1 г лигнина тополя.Полученный лигнин растворяли в 550 мкл диметилсульфоксида (ДМСО)-d6 и охарактеризовали с помощью 1 H/ 13 C двумерного гетероядерного одноквантового когерентного (HSQC) ЯМР и количественного анализа 13 C ЯМР с использованием Bruker Avance III.
После этого жидкость концентрировали под вакуумом при 30°С до помутнения жидкости и снова растворяли в 30 мл ацетона. Затем концентрированный раствор лигнина медленно вливали в быстро перемешиваемую 2000 мл воды и отфильтровывали осадок с помощью воронки с размером пор 3–4 мкм.Затем лигнин-сырец растворяли в 150 мл ацетона/H 2 O (9:1) и снова осаждали в воду тем же способом. Наконец, сырой лигнин растворяют в 200 мл раствора ацетон/метанол (9:1) и сушат над безводным MgSO 4 . Затем жидкость медленно вливали в быстро перемешиваемые 2000 мл диэтилового эфира и фильтровали с использованием воронки с размером пор от 3 до 4 мкм. Твердое вещество лигнина лиофилизировали в вакууме в течение 24 часов и в итоге получили 3,1 г лигнина тополя.Полученный лигнин растворяли в 550 мкл диметилсульфоксида (ДМСО)-d6 и охарактеризовали с помощью 1 H/ 13 C двумерного гетероядерного одноквантового когерентного (HSQC) ЯМР и количественного анализа 13 C ЯМР с использованием Bruker Avance III.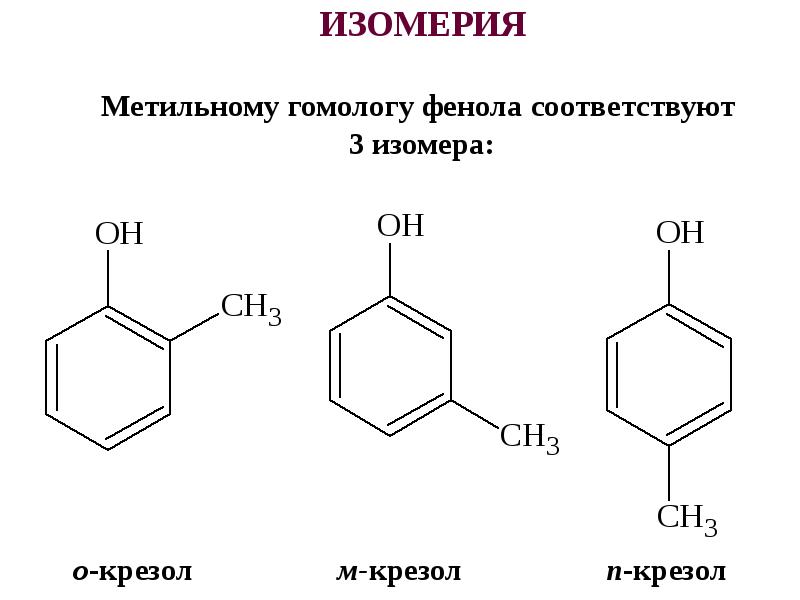 500WB и Bruker Avance 600, как описано другими исследователями ( 33 ). Для количественного анализа ЯМР 13 С в качестве релаксационного реагента использовали ацетилацетонат хрома(III).
500WB и Bruker Avance 600, как описано другими исследователями ( 33 ). Для количественного анализа ЯМР 13 С в качестве релаксационного реагента использовали ацетилацетонат хрома(III). Синтез модельных соединений
1 Н ЯМР и 13 С ЯМР анализы выполняли на Bruker Avance III 400 HD с использованием ДМСО-d6 в качестве растворителя.Химические сдвиги 1 H относятся к ТМС при 0 м.д., а химические сдвиги 13 C относятся к ДМСО-d6 при 39,6 м.д. Множественности описываются с использованием следующих сокращений: с, синглет; д, дублет; т, триплет; кв, квартет; и м, мультиплет. Подробности методов и процедур синтеза модельных соединений описаны в дополнительном тексте.
Каталитическая характеристика
Реакции проводили в реакторе из нержавеющей стали с тефлоновым покрытием объемом 20 мл с магнитной мешалкой.В типичном эксперименте в реактор загружали подходящее количество реагента, катализатора и воды. Реактор герметизировали и трижды продували аргоном для удаления воздуха при комнатной температуре, а затем загружали 0,5 МПа аргона. Затем реактор помещали в печь при желаемой температуре реакции. Затем включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. После реакции реактор помещали в воду со льдом, и газ выпускали, пропуская через этилацетат.Реакционную смесь в реакторе переносили в центрифужную пробирку. Затем реактор промывали этилацетатом, использованным для фильтрации газа, который в последнюю очередь смешивали с реакционной смесью. После центрифугирования катализатор отделяли от реакционной смеси. Количественный анализ жидких продуктов в органической фазе проводили с помощью ГХ (Agilent 6820), оснащенного пламенно-ионизационным детектором и капиллярной колонкой HP-5ms (диаметр 0,25 мм, длина 30 м).Идентификацию продуктов и реагентов проводили с помощью системы ГХ-масс-спектрометрии (ГХ-МС) [Agilent 5977A, капиллярная колонка HP-5ms (0,25 м в диаметре и 30 м в длину)] и путем сравнения времени удерживания с соответствующими стандарты в трассировках GC. Бифенил использовали в качестве внутреннего стандарта для определения конверсии субстратов, селективности и выхода продуктов.
Затем реактор помещали в печь при желаемой температуре реакции. Затем включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. После реакции реактор помещали в воду со льдом, и газ выпускали, пропуская через этилацетат.Реакционную смесь в реакторе переносили в центрифужную пробирку. Затем реактор промывали этилацетатом, использованным для фильтрации газа, который в последнюю очередь смешивали с реакционной смесью. После центрифугирования катализатор отделяли от реакционной смеси. Количественный анализ жидких продуктов в органической фазе проводили с помощью ГХ (Agilent 6820), оснащенного пламенно-ионизационным детектором и капиллярной колонкой HP-5ms (диаметр 0,25 мм, длина 30 м).Идентификацию продуктов и реагентов проводили с помощью системы ГХ-масс-спектрометрии (ГХ-МС) [Agilent 5977A, капиллярная колонка HP-5ms (0,25 м в диаметре и 30 м в длину)] и путем сравнения времени удерживания с соответствующими стандарты в трассировках GC. Бифенил использовали в качестве внутреннего стандарта для определения конверсии субстратов, селективности и выхода продуктов.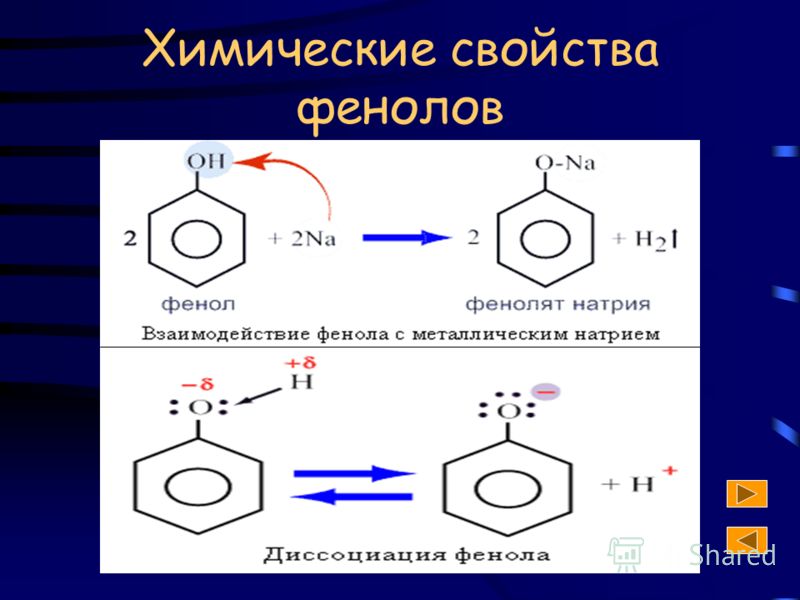 Углеродный баланс для превращения модельных соединений рассчитывали, используя балансы C ароматических соединений , которые даны относительно ароматических продуктов ( 50 ).Баланс ароматических соединений C для превращения модельных соединений превышал 95%. Баланс массы для превращения лигнина рассчитывали по уравнению 1 ( 35 ). После реакции твердое вещество и жидкость разделяли центрифугированием, и масса извлеченного твердого вещества была почти такой же, как масса загруженного катализатора. Для трансформации лигнина использовали внутренний стандарт (бифенил для органической фазы) для определения выхода обнаруживаемых продуктов. После извлечения катализатора жидкие реакционные смеси подвергали ротационному испарителю для удаления растворителей и летучих продуктов.Затем извлеченное остаточное твердое вещество подвергали лиофилизации в вакууме в течение 24 часов. Для превращения лигнина, катализируемого цеолитом HY 30 , массовый баланс составил 90 ± 5%.
Углеродный баланс для превращения модельных соединений рассчитывали, используя балансы C ароматических соединений , которые даны относительно ароматических продуктов ( 50 ).Баланс ароматических соединений C для превращения модельных соединений превышал 95%. Баланс массы для превращения лигнина рассчитывали по уравнению 1 ( 35 ). После реакции твердое вещество и жидкость разделяли центрифугированием, и масса извлеченного твердого вещества была почти такой же, как масса загруженного катализатора. Для трансформации лигнина использовали внутренний стандарт (бифенил для органической фазы) для определения выхода обнаруживаемых продуктов. После извлечения катализатора жидкие реакционные смеси подвергали ротационному испарителю для удаления растворителей и летучих продуктов.Затем извлеченное остаточное твердое вещество подвергали лиофилизации в вакууме в течение 24 часов. Для превращения лигнина, катализируемого цеолитом HY 30 , массовый баланс составил 90 ± 5%.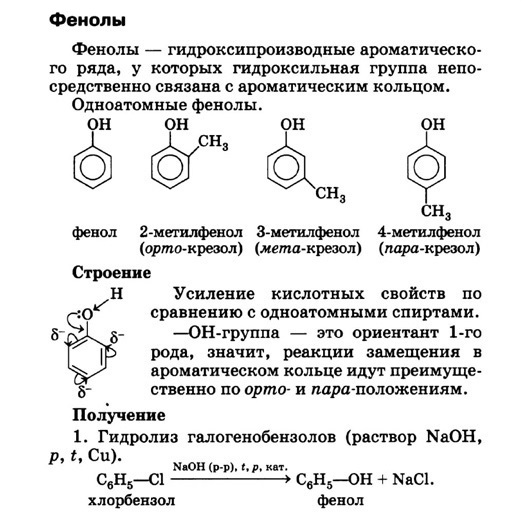 Структуру лигнина и извлеченного остаточного твердого вещества анализировали с помощью 1 H/ 13 C 2D HSQC ЯМР и количественной 13 C ЯМР-спектроскопии (Bruker Avance III 600 HD). 100%
Структуру лигнина и извлеченного остаточного твердого вещества анализировали с помощью 1 H/ 13 C 2D HSQC ЯМР и количественной 13 C ЯМР-спектроскопии (Bruker Avance III 600 HD). 100% (1)
Повторное использование катализатора
Возможность повторного использования цеолита HY 30 была протестирована с использованием реакции модельного соединения 1a .После реакции реакционную смесь в реакторе переносили в центрифужную пробирку. Затем реактор промывали этилацетатом, который в последнюю очередь смешивали с реакционной смесью. Использованный цеолит HY 30 после центрифугирования отделяли от реакционной смеси и последовательно промывали этанолом (5 х 10 мл) и водой (5 х 10 мл). Затем извлеченный цеолит сушили при 120°С в течение 2 часов и затем прокаливали при 550°С в течение 4 часов. После прокаливания рециркулированный цеолит повторно использовали непосредственно для следующего запуска.
Масштабирование лигнина тополя
Реакцию масштабирования проводили в 1-литровом автоклаве (Weihai Xinyuan Chemical Machinery Co.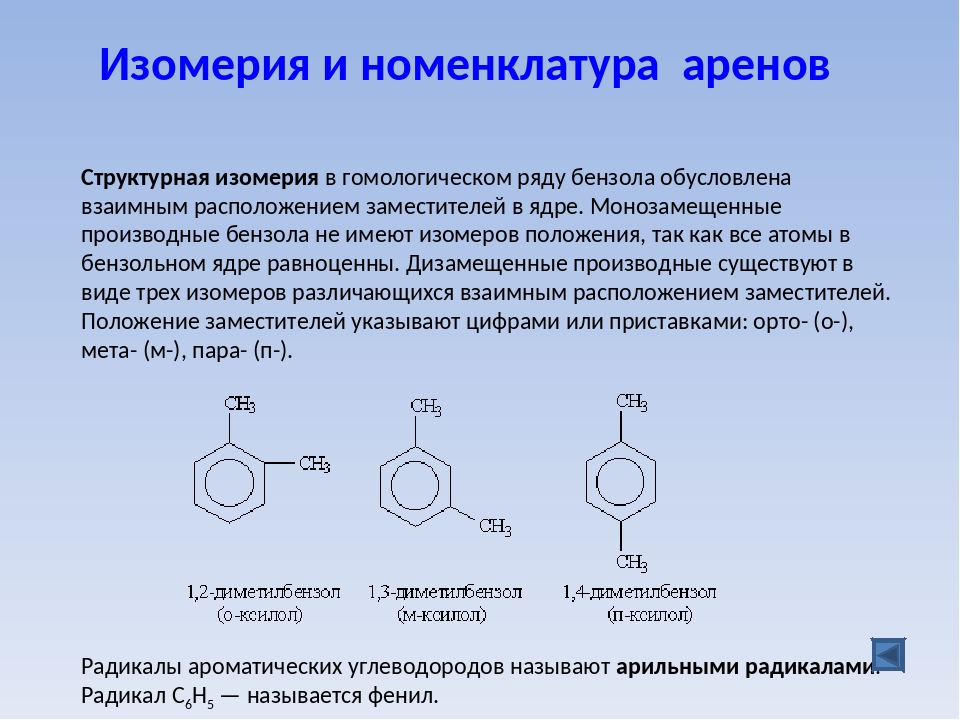 Ltd.). В реакционную смесь в автоклав добавляли 50,0 г лигнина, 50,0 г HY 30 и 600 мл H 2 O. Автоклав герметизировали и продували аргоном для удаления воздуха при комнатной температуре, а затем загружали аргоном под давлением 0,5 МПа. Затем автоклав нагревали до 200°С, и в течение 40 мин автоклав достигал желаемой температуры реакции.После этого включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. После реакции автоклав охлаждали до комнатной температуры и выпускали газ. Жидкие продукты экстрагировали из реакционной смеси этилацетатом и концентрировали в вакууме. Затем сырой фенол очищали на колонке с силикагелем, используя в качестве растворителя смесь этилацетат–петролейный эфир (1:4, по объему), и концентрировали в вакууме. Наконец, в процессе кристаллизации был получен чистый фенол.
Ltd.). В реакционную смесь в автоклав добавляли 50,0 г лигнина, 50,0 г HY 30 и 600 мл H 2 O. Автоклав герметизировали и продували аргоном для удаления воздуха при комнатной температуре, а затем загружали аргоном под давлением 0,5 МПа. Затем автоклав нагревали до 200°С, и в течение 40 мин автоклав достигал желаемой температуры реакции.После этого включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. После реакции автоклав охлаждали до комнатной температуры и выпускали газ. Жидкие продукты экстрагировали из реакционной смеси этилацетатом и концентрировали в вакууме. Затем сырой фенол очищали на колонке с силикагелем, используя в качестве растворителя смесь этилацетат–петролейный эфир (1:4, по объему), и концентрировали в вакууме. Наконец, в процессе кристаллизации был получен чистый фенол.
18 Испытание мечения изотопов O
Эксперимент по мечению изотопов проводили в реакторе из нержавеющей стали с тефлоновым покрытием объемом 20 мл с магнитной мешалкой. В эксперименте в реактор загружали 4-(1-гидроксипропил)фенол (1а) (1 ммоль), HY 30 (0,3 г) и H 2 18 O (4,0 мл). Реактор герметизировали и трижды продували аргоном для удаления воздуха при комнатной температуре, а затем загружали 0,5 МПа аргона. Затем реактор помещали в печь и нагревали до 180°С.Затем включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. Через 1 час реактор помещали в ледяную воду и выпускали газ. Реакционную смесь в реакторе переносили в центрифужную пробирку. Затем реактор промывали этилацетатом. После центрифугирования катализатор отделяли от реакционной смеси. Идентификацию продуктов проводили с использованием ГХ-МС [Agilent 5977A, капиллярная колонка HP-5ms (диаметр 0,25 нм и длина 30 м)] путем сравнения спектров МС продуктов со спектрами сравнения.
В эксперименте в реактор загружали 4-(1-гидроксипропил)фенол (1а) (1 ммоль), HY 30 (0,3 г) и H 2 18 O (4,0 мл). Реактор герметизировали и трижды продували аргоном для удаления воздуха при комнатной температуре, а затем загружали 0,5 МПа аргона. Затем реактор помещали в печь и нагревали до 180°С.Затем включали мешалку со скоростью перемешивания 800 об/мин и регистрировали время реакции. Через 1 час реактор помещали в ледяную воду и выпускали газ. Реакционную смесь в реакторе переносили в центрифужную пробирку. Затем реактор промывали этилацетатом. После центрифугирования катализатор отделяли от реакционной смеси. Идентификацию продуктов проводили с использованием ГХ-МС [Agilent 5977A, капиллярная колонка HP-5ms (диаметр 0,25 нм и длина 30 м)] путем сравнения спектров МС продуктов со спектрами сравнения.
Обнаружение промежуточных продуктов
Обнаружение промежуточных продуктов проводили в реакторе из нержавеющей стали с тефлоновым покрытием объемом 20 мл с магнитной мешалкой.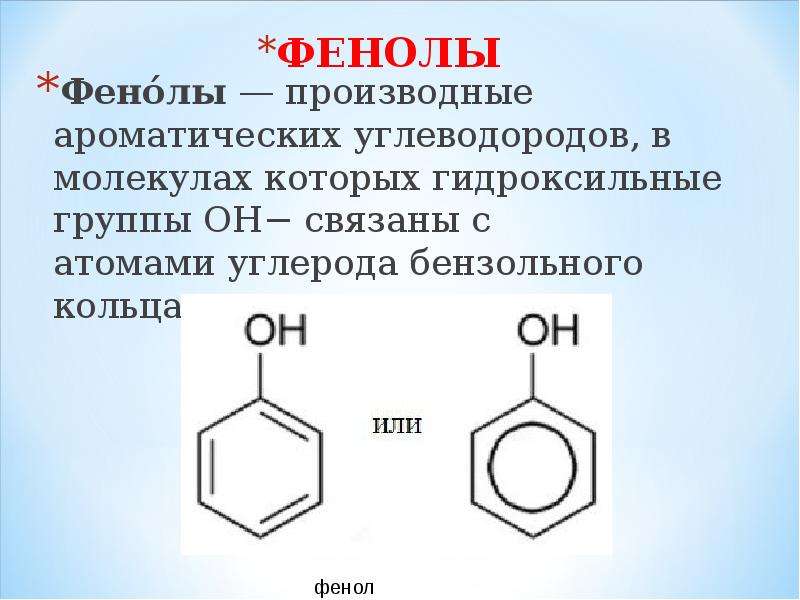 В эксперименте в реактор загружали 1-(4-метоксифенил)пропан-1-ол (1а) (1 ммоль), HY 30 (0,3 г) и H 2 O (4,0 мл). Реактор герметизировали и трижды продували аргоном для удаления воздуха при комнатной температуре, а затем загружали 0,5 МПа аргона. Затем реактор помещали в печь и нагревали до 180°С, включали мешалку со скоростью перемешивания 800 об/мин и фиксировали время реакции.Через 10 мин реактор очень быстро помещали в ванну с жидким азотом. При замораживании реакционной смеси газ сразу выпускали. Реакционную смесь лиофилизировали в вакууме в течение 12 часов для удаления воды. Обнаружение промежуточных соединений в высушенном образце проводили с помощью 13 C ЯМР, твердотельного 2D 13 C { 1 H} диполярно-опосредованного HETCOR и 27 Al ЯМР анализов на Bruker Avance III 400. Твердотельный 2D 13 C{ 1 H} эксперимент HETCOR был проведен в соответствии с процедурой, описанной McGehee и соавторами ( 51 ).
В эксперименте в реактор загружали 1-(4-метоксифенил)пропан-1-ол (1а) (1 ммоль), HY 30 (0,3 г) и H 2 O (4,0 мл). Реактор герметизировали и трижды продували аргоном для удаления воздуха при комнатной температуре, а затем загружали 0,5 МПа аргона. Затем реактор помещали в печь и нагревали до 180°С, включали мешалку со скоростью перемешивания 800 об/мин и фиксировали время реакции.Через 10 мин реактор очень быстро помещали в ванну с жидким азотом. При замораживании реакционной смеси газ сразу выпускали. Реакционную смесь лиофилизировали в вакууме в течение 12 часов для удаления воды. Обнаружение промежуточных соединений в высушенном образце проводили с помощью 13 C ЯМР, твердотельного 2D 13 C { 1 H} диполярно-опосредованного HETCOR и 27 Al ЯМР анализов на Bruker Avance III 400. Твердотельный 2D 13 C{ 1 H} эксперимент HETCOR был проведен в соответствии с процедурой, описанной McGehee и соавторами ( 51 ).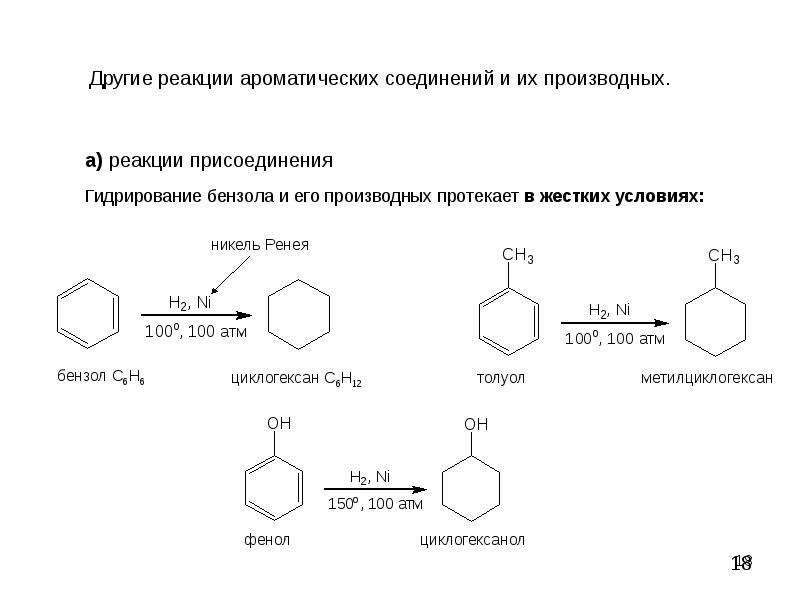
Вычислительная методология
Программный пакет моделирования Vienna ab initio, основанный на методе расширенных волн с замороженным ядром для полностью электронного проектора ( 52 ) и основе плоских волн, использовался для выполнения спин-поляризованных вычислений DFT. Использовался функционал DFT PBE-D3 (функционал Perdew, Burke и Ernzerhof с последней поправкой на дисперсию). Это соответствует функционалу PBE и поправке на дисперсионные эффекты, предложенные Гриммом и др. . (ДПФ-D3) ( 53 ).Было выбрано значение отсечки конвергентной кинетической энергии 500 эВ. Оптимизация структуры проводилась путем минимизации сил с помощью алгоритма сопряженных градиентов до тех пор, пока сила, действующая на каждый ион, не станет ниже 0,02 эВ/Å, а критерии сходимости для электронных самосогласованных взаимодействий составят 10 -5 . Поправка на нулевую колебательную энергию не учитывалась. Путь минимальной энергии для элементарных стадий реакции был выполнен с использованием расчета NEB (смещенной эластичной ленты) с изображением восхождения ( 54 ).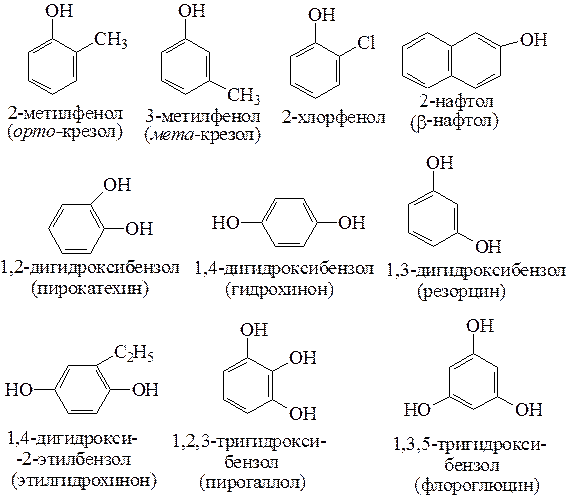
Низкосимметричная ромбоэдрическая элементарная ячейка использовалась для моделирования фожазитового цеолита (a = b = c = 12,22 Å, α = β = γ = 60°). Общая формула фожазитового цеолита с компонентом чистого кремния: Si 48 O 96 . Каркас фожазитового цеолита содержит только одну кристаллографически тетраэдрическую Т-позицию. На основании наших экспериментальных результатов цеолиты HY с соотношением Si/Al, равным 30, продемонстрировали наилучшие каталитические характеристики, указывая на то, что в элементарной ячейке имеется приблизительно два атома алюминия.Поэтому в нашу модель элементарной ячейки HY были включены два атома алюминия. Кроме того, разумно предположить, что два атома алюминия в элементарной ячейке пространственно соседствуют, т. е. одна из позиций используется как активная, а другая из-за нехватки места может адсорбировать или десорбировать молекулы воды, способствуя Реакция.
Энергию реакции (Δ E ) и энергию активации (Ea) рассчитывали по следующим двум формулам: Δ E = E(FS) − E (IS) и Ea = E (TS) − E (ИС) соответственно. Здесь E (TS), E (IS) и E (FS) — расчетные энергии переходного состояния, начального состояния и конечного состояния каждой элементарной ступени соответственно.
Здесь E (TS), E (IS) и E (FS) — расчетные энергии переходного состояния, начального состояния и конечного состояния каждой элементарной ступени соответственно.
 энтропийная энергия 79 и 0,83 эВ соответственно.
энтропийная энергия 79 и 0,83 эВ соответственно. Неожиданное превращение растворенных фенолов в токсичные дикарбонилы под действием гидроксильных радикалов и УФ-излучения
К 2050 году две трети населения мира будут проживать в городах, которые все больше зависят от источников питьевой воды, подверженных воздействию сельскохозяйственных стоков, а также промышленных и городских сточных вод. (1, 2). Эти источники питьевой воды часто содержат следовые концентрации фенольных соединений, которые широко используются в красителях, поверхностно-активных веществах, фармацевтических препаратах и пестицидах, включая бисфенол А, триклозан и нонилфенолэтоксилаты (3, 4).В результате опасений по поводу неблагоприятных последствий для здоровья от хронического воздействия фенольных соединений Агентство по охране окружающей среды и другие регулирующие органы требуют удаления определенных фенольных соединений при очистке воды. Один из подходов к обработке фенолсодержащей воды включает окисление этих соединений гидроксильными радикалами (•ОН).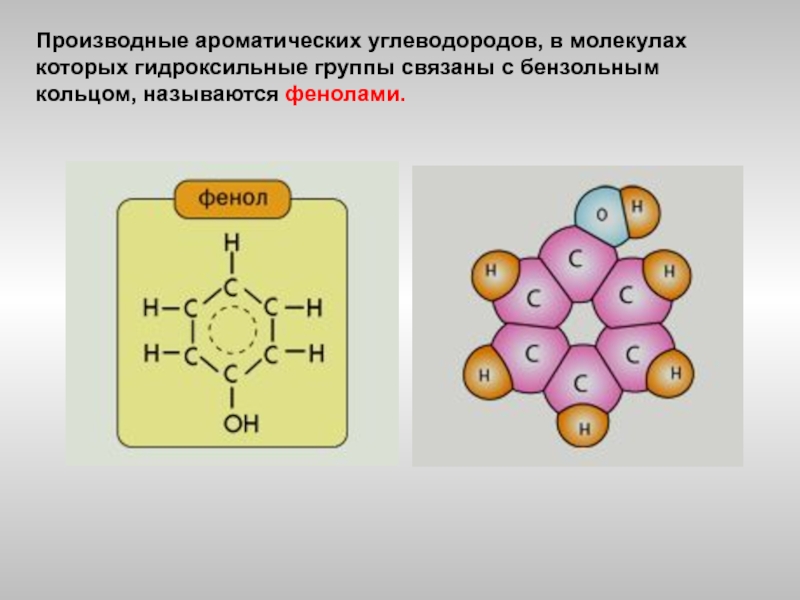 • Технологии очистки на основе OH, такие как использование УФ-излучения для фотолиза перекиси водорода (т. е. процесс UV/H 2 O 2 ), становятся все более распространенными при очистке питьевой воды, повторном использовании питьевой воды и восстановлении. загрязненных подземных вод на полигонах опасных отходов.В этих процессах •ОН трансформирует фенольные соединения и другие органические загрязнители посредством ряда реакций, которые приводят к присоединению кислородсодержащих функциональных групп к соединениям (5, 6). Хотя продукты превращения, образующиеся в этих реакциях, зачастую менее токсичны, чем исходные соединения, и часто могут быть легче удалены в последующих процессах очистки воды, окисление фенолов также может привести к образованию токсичных продуктов превращения, таких как р- и o -бензохинон (7).Воздействие хинонов вызывает токсикологическую озабоченность, поскольку их электрофильный характер приводит к повреждению клеток в результате реакций с нуклеофильными группами в белках и ДНК (8).
• Технологии очистки на основе OH, такие как использование УФ-излучения для фотолиза перекиси водорода (т. е. процесс UV/H 2 O 2 ), становятся все более распространенными при очистке питьевой воды, повторном использовании питьевой воды и восстановлении. загрязненных подземных вод на полигонах опасных отходов.В этих процессах •ОН трансформирует фенольные соединения и другие органические загрязнители посредством ряда реакций, которые приводят к присоединению кислородсодержащих функциональных групп к соединениям (5, 6). Хотя продукты превращения, образующиеся в этих реакциях, зачастую менее токсичны, чем исходные соединения, и часто могут быть легче удалены в последующих процессах очистки воды, окисление фенолов также может привести к образованию токсичных продуктов превращения, таких как р- и o -бензохинон (7).Воздействие хинонов вызывает токсикологическую озабоченность, поскольку их электрофильный характер приводит к повреждению клеток в результате реакций с нуклеофильными группами в белках и ДНК (8).
Несмотря на растущее признание того, что при окислительной очистке воды могут образовываться токсичные продукты трансформации (9), потенциальные последствия этой практики для здоровья неясны. Поскольку в источниках питьевой воды могут присутствовать тысячи антропогенных соединений, оценка воздействия каждого соединения, которое может присутствовать, не представляется возможным.Скорее, необходимы новые подходы для определения приоритетности дальнейших исследований соединений, которые являются токсичными по своей природе или которые могут быть преобразованы в токсичные продукты трансформации во время очистки воды.
В токсикологии признание важности молекулярных взаимодействий химических веществ с биомолекулами привело к разработке концепции пути неблагоприятного исхода (10). В качестве ключевой особенности молекулярные инициирующие события (например, образование ковалентных аддуктов в результате реакции как эндогенных, так и экзогенных электрофилов с белками и ДНК) были признаны важным механизмом, участвующим в различных неблагоприятных последствиях для здоровья, включая рак и сердечно-сосудистые заболевания.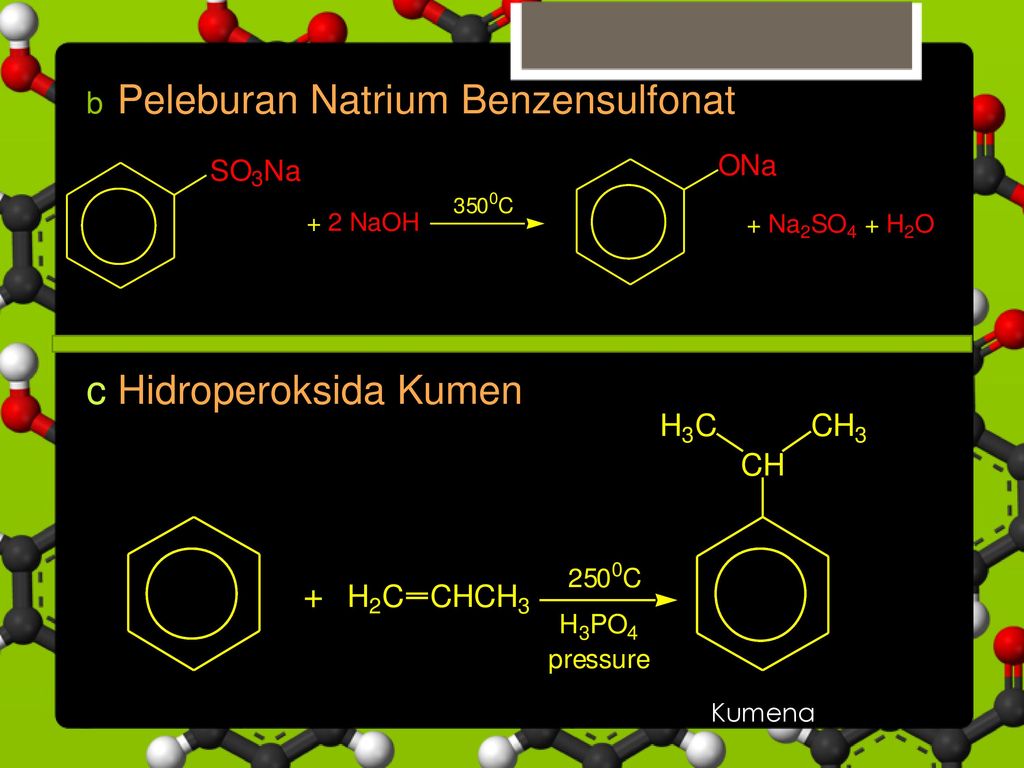 (11, 12).Это также привело к разработке инструментов скрининга, которые позволяют оценивать реактивные кандидаты в фармацевтические препараты и их метаболиты путем исследования образования ковалентных аддуктов, образующихся при взаимодействии тестируемых соединений с аминокислотами и белками (13, 14). Чтобы оценить потенциал токсичных продуктов окислительной обработки воды, мы адаптировали этот подход для выявления реактивных электрофилов, которые образуются при окислительной очистке воды. Чтобы обеспечить чувствительное обнаружение продуктов окисления, имеющих токсикологическую значимость, мы нацелились на аддукты, образующиеся при взаимодействии продуктов окисления с нуклеофильными фрагментами в биомолекулах, включая фрагменты первичных аминов в N -α-ацетиллизине (NAL) и тиоловые функциональные группы в глутатионе. (GSH) и N -ацетилцистеин (NAC).
(11, 12).Это также привело к разработке инструментов скрининга, которые позволяют оценивать реактивные кандидаты в фармацевтические препараты и их метаболиты путем исследования образования ковалентных аддуктов, образующихся при взаимодействии тестируемых соединений с аминокислотами и белками (13, 14). Чтобы оценить потенциал токсичных продуктов окислительной обработки воды, мы адаптировали этот подход для выявления реактивных электрофилов, которые образуются при окислительной очистке воды. Чтобы обеспечить чувствительное обнаружение продуктов окисления, имеющих токсикологическую значимость, мы нацелились на аддукты, образующиеся при взаимодействии продуктов окисления с нуклеофильными фрагментами в биомолекулах, включая фрагменты первичных аминов в N -α-ацетиллизине (NAL) и тиоловые функциональные группы в глутатионе. (GSH) и N -ацетилцистеин (NAC).
Результаты и обсуждение
Для оценки репрезентативной системы очистки мы окислили фенол с помощью •OH, полученного в процессе УФ/Н 2 O 2 . После воздействия мы добавили NAL и обнаружили образование аддукта R-2-(ацетиламино)-6-(2,5-дигидро-2-оксо-1H-пиррол-1-ил)-1-гексановой кислоты (). m / z 255), концентрация которых увеличивалась по мере снижения концентрации фенола (рис. 1 A и SI Приложение , рис. S1). Аналогичным образом добавление либо GSH, либо эквимолярной смеси NAC и NAL после окисления фенола в процессе UV/H 2 O 2 дает аддукты N -[4-карбокси-4-(3-меркапто- 1H-пиррол-1-ил)-1-оксобутил]-1-цистеинилглицин циклический сульфид ( m / z 356) и N -ацетил-S-[1-[5-(ацетиламино)-5- аддукты карбоксипентил]-1H-пиррол-3-ил]-1-цистеина ( m / z 400) соответственно.Эти данные показывают, что окисление фенола •ОН приводит к образованию α,β-ненасыщенного диальдегида 2-бутен-1,4-диал (15). Образование того же N -замещенного пирролин-2-она и аддуктов пиррола наблюдалось ранее во время CYP450-опосредованного метаболизма фурана, соединения, которое обычно содержится в термически обработанных пищевых продуктах, сигаретном дыме и выхлопных газах.
После воздействия мы добавили NAL и обнаружили образование аддукта R-2-(ацетиламино)-6-(2,5-дигидро-2-оксо-1H-пиррол-1-ил)-1-гексановой кислоты (). m / z 255), концентрация которых увеличивалась по мере снижения концентрации фенола (рис. 1 A и SI Приложение , рис. S1). Аналогичным образом добавление либо GSH, либо эквимолярной смеси NAC и NAL после окисления фенола в процессе UV/H 2 O 2 дает аддукты N -[4-карбокси-4-(3-меркапто- 1H-пиррол-1-ил)-1-оксобутил]-1-цистеинилглицин циклический сульфид ( m / z 356) и N -ацетил-S-[1-[5-(ацетиламино)-5- аддукты карбоксипентил]-1H-пиррол-3-ил]-1-цистеина ( m / z 400) соответственно.Эти данные показывают, что окисление фенола •ОН приводит к образованию α,β-ненасыщенного диальдегида 2-бутен-1,4-диал (15). Образование того же N -замещенного пирролин-2-она и аддуктов пиррола наблюдалось ранее во время CYP450-опосредованного метаболизма фурана, соединения, которое обычно содержится в термически обработанных пищевых продуктах, сигаретном дыме и выхлопных газах.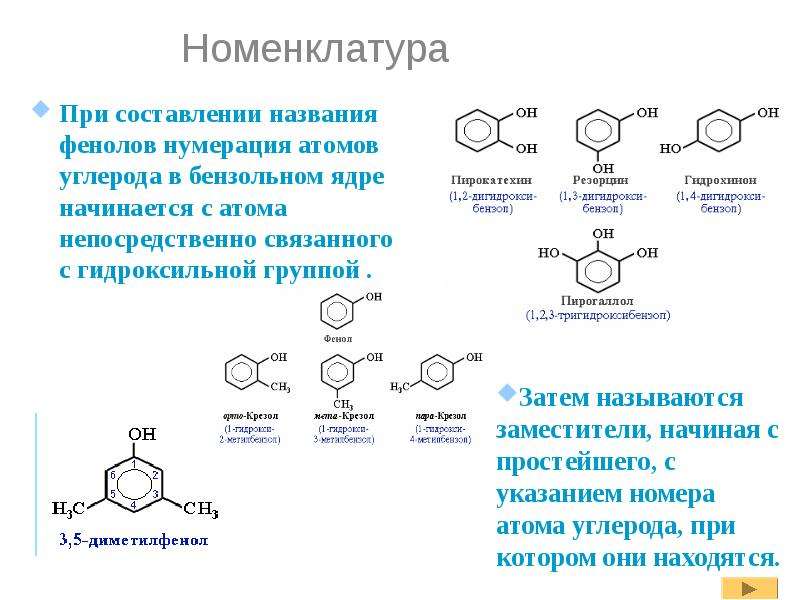 автомобильные и дизельные двигатели (рис. 1 B ) (16, 17). 2-бутен-1,4-диал реагирует с белковыми и ДНК-нуклеофилами, демонстрирует сильный положительный ответ в анализе Эймса, вызывает разрывы цепей и перекрестные связи в ДНК и связан с токсичностью фурана in vivo (16, 18).
автомобильные и дизельные двигатели (рис. 1 B ) (16, 17). 2-бутен-1,4-диал реагирует с белковыми и ДНК-нуклеофилами, демонстрирует сильный положительный ответ в анализе Эймса, вызывает разрывы цепей и перекрестные связи в ДНК и связан с токсичностью фурана in vivo (16, 18).
Образование 2-бутен-1,4-диала при окислении фенола в процессе УФ/Н 2 O 2 . ( A ) Окисление фенола (0,1 мМ) в боратном буфере (50 мМ при pH 8) с помощью УФ/Н 2 O 2 и реакция 2-бутен-1,4-диала с N -α-ацетил-1-лизин (NAL), глутатион (GSH) или смесь NAL и NAC. В качестве источника излучения использовалась ртутная лампа среднего давления. ( B ) Образование 2-бутен-1,4-диала в результате реакции фенола с •OH и УФ-светом или путем биоактивации фурана цитохромом P450 (CYP450) в организме человека (33) и образование аддукта с NAL, GSH, а также NAC и NAL.( C ) Окисление фенола УФ-светом в отсутствие (сплошные треугольники) и в присутствии H 2 O 2 (0,1 мМ; сплошные кружки) и количественное определение 2-бутен-1,4-диала с помощью ЖХ /МС/МС с использованием стандартного метода сложения (незаштрихованные кружки и треугольники; см. Приложение SI ).
Приложение SI ).
Количественный анализ 2-бутен-1,4-диал с помощью ЖХ/МС/МС показал, что ~2% потерянного фенола превратилось в α,β-ненасыщенный диальдегид (рис. 1 C ) .В отдельном эксперименте мы также продемонстрировали, что 2-бутен-1,4-диал образуется, хотя и с меньшими выходами, при прямом УФ-фотолизе фенола ртутной лампой среднего давления (рис. 1 C ). Воздействие на фенол света с длиной волны выше 290 нм не приводило к разложению фенола или образованию 2-бутен-1,4-диала. Образование продукта превращения наблюдалось и при использовании в качестве источника света ртутной лампы низкого давления.
Десятилетия исследований установили путь окисления фенола с помощью •OH или УФ-излучения, состоящий из начального гидроксилирования ароматического кольца с образованием гидрохинона, бензохинона и катехола с последующим гидроксилированием и расщеплением кольца с образованием органических кислот с короткой цепью. , такие как малеиновая кислота, муравьиная кислота и щавелевая кислота (7).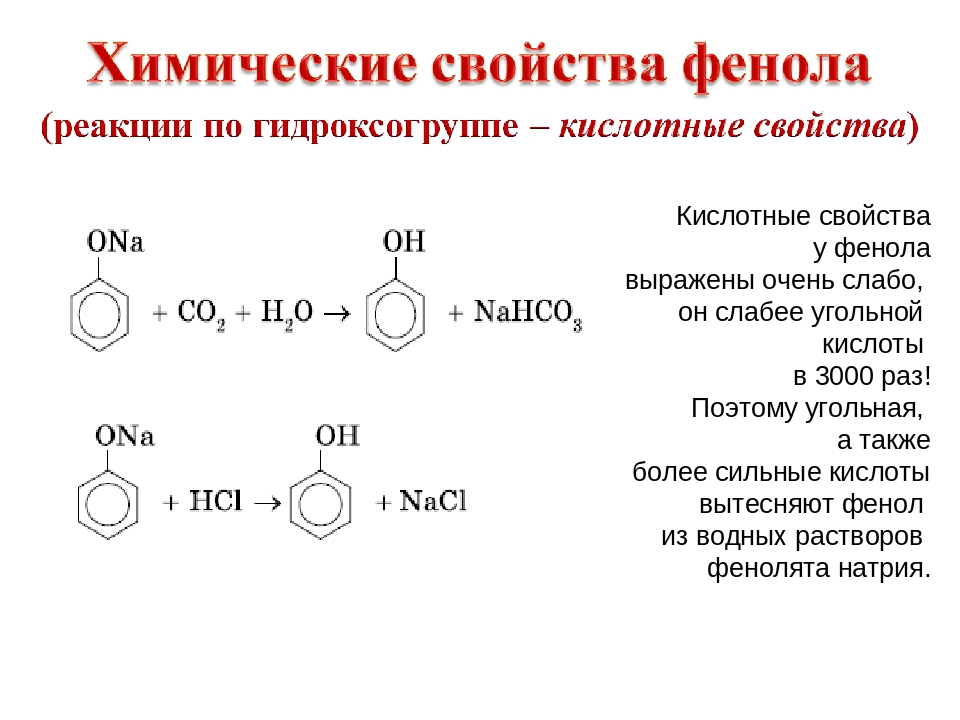 Было постулировано прямое расщепление ароматических колец без последовательного гидроксилирования (19), но наши результаты представляют собой экспериментальное свидетельство того, что α,β-ненасыщенные альдегиды образуются как исходный продукт окисления фенола в водной фазе под воздействием • ОН или УФ-излучения. В газовой фазе (20 ⇓⇓⇓–24). На начальном этапе механизма газофазной реакции образуются углерод-центрированные радикалы либо путем отщепления H, либо путем добавления OH, которые впоследствии реагируют с молекулярным кислородом с образованием пероксидов.Эти нестабильные промежуточные соединения могут подвергаться мономолекулярному замыканию кольца с образованием бициклических алкоксирадикалов, которые затем распадаются с образованием энедиалов и оксеналей. На этом последнем этапе исходные шесть атомов углерода ароматического кольца превращаются в четырехуглеродное и двухуглеродное соединение. В водной фазе постулируется образование бициклических алкоксирадикалов после реакции •ОН с бензолом и фенолом в водной фазе (25, 26), но наши результаты экспериментально свидетельствуют о протекании этого процесса в воде.
Было постулировано прямое расщепление ароматических колец без последовательного гидроксилирования (19), но наши результаты представляют собой экспериментальное свидетельство того, что α,β-ненасыщенные альдегиды образуются как исходный продукт окисления фенола в водной фазе под воздействием • ОН или УФ-излучения. В газовой фазе (20 ⇓⇓⇓–24). На начальном этапе механизма газофазной реакции образуются углерод-центрированные радикалы либо путем отщепления H, либо путем добавления OH, которые впоследствии реагируют с молекулярным кислородом с образованием пероксидов.Эти нестабильные промежуточные соединения могут подвергаться мономолекулярному замыканию кольца с образованием бициклических алкоксирадикалов, которые затем распадаются с образованием энедиалов и оксеналей. На этом последнем этапе исходные шесть атомов углерода ароматического кольца превращаются в четырехуглеродное и двухуглеродное соединение. В водной фазе постулируется образование бициклических алкоксирадикалов после реакции •ОН с бензолом и фенолом в водной фазе (25, 26), но наши результаты экспериментально свидетельствуют о протекании этого процесса в воде.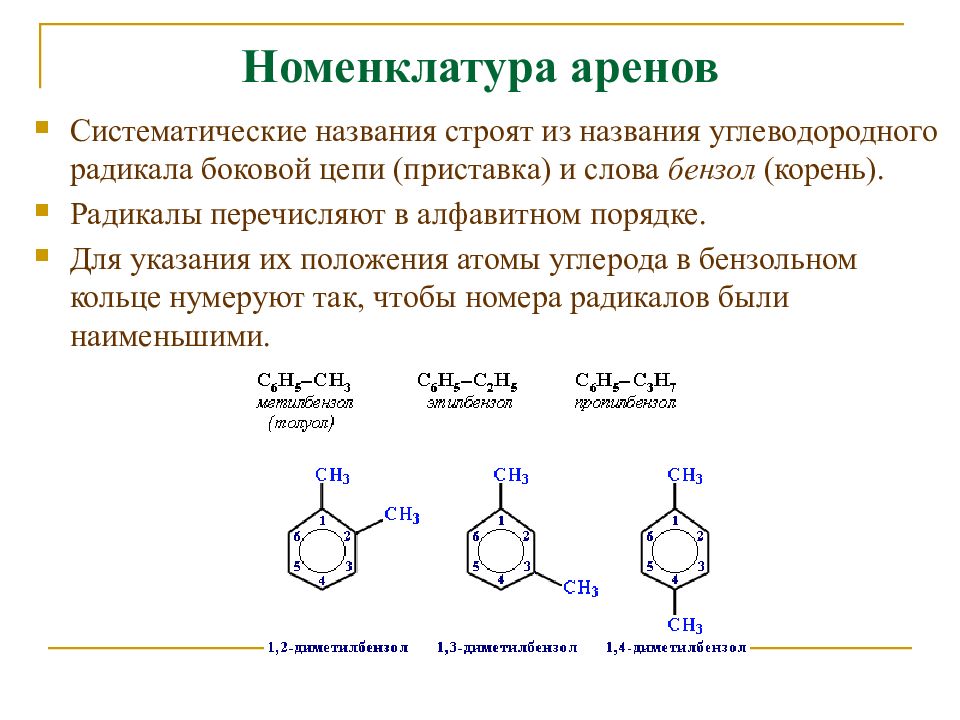
Токсикологи нашли убедительные доказательства того, что 2-бутен-1,4-диал, образующийся в результате метаболизма фурана, вызывает токсичность и гибель клеток за счет связывания с печеночными белками (27). 2-бутен-1,4-диал представляет собой высокореакционноспособный электрофил, который потенциально может реагировать с цистеинами в белках. Чтобы лучше понять потенциальную токсичность и реактивность цистеина в масштабах всего протеома 2-бутен-1,4-диала, мы использовали хемопротеомную платформу, называемую профилированием белка на основе активности (ABPP). ABPP использует зонды, основанные на реактивности, для картирования реактивных, функциональных и лигандируемых «горячих точек» всего протеома непосредственно в сложных протеомах (28, 29).Чтобы первоначально определить, обладает ли 2-бутен-1,4-диал какой-либо цистеиновой реактивностью с белками в протеомах печени мышей, мы провели анализ ABPP в геле, в котором мы сравнили его связывание с белками со связыванием цистеин-реактивного йодацетамидного алкина. (IAyne) зонд (30). Мы обнаружили, что 2-бутен-1,4-диал проявляет широкую реакционную способность по отношению к цистеину в зависимости от дозы, демонстрируя более селективную реактивность с белками при более низких концентрациях (рис. 2 A ). Для картирования специфических лигандируемых цистеиновых горячих точек, на которые нацелен 2-бутен-1,4-диал, мы использовали более совершенную платформу ABPP, называемую ABPP с изотопным тандемным ортогональным протеолизом (isoTOP-ABPP) (28).В этом методе используется биотин-азидная метка, содержащая изотопно легкую (для обработки носителем) или тяжелую (для обработки 1,4-бутендиалом) метку и последовательность распознавания протеазы вируса травления табака (TEV), которая связана с IAyne- меченые белки с помощью клик-химии. Таким образом, он позволяет проводить количественный протеомный анализ с использованием соотношений легких и тяжелых пептидов, модифицированных зондом. Мы интерпретировали эти пептиды, показывающие соотношение >10, как прямые мишени.
(IAyne) зонд (30). Мы обнаружили, что 2-бутен-1,4-диал проявляет широкую реакционную способность по отношению к цистеину в зависимости от дозы, демонстрируя более селективную реактивность с белками при более низких концентрациях (рис. 2 A ). Для картирования специфических лигандируемых цистеиновых горячих точек, на которые нацелен 2-бутен-1,4-диал, мы использовали более совершенную платформу ABPP, называемую ABPP с изотопным тандемным ортогональным протеолизом (isoTOP-ABPP) (28).В этом методе используется биотин-азидная метка, содержащая изотопно легкую (для обработки носителем) или тяжелую (для обработки 1,4-бутендиалом) метку и последовательность распознавания протеазы вируса травления табака (TEV), которая связана с IAyne- меченые белки с помощью клик-химии. Таким образом, он позволяет проводить количественный протеомный анализ с использованием соотношений легких и тяжелых пептидов, модифицированных зондом. Мы интерпретировали эти пептиды, показывающие соотношение >10, как прямые мишени.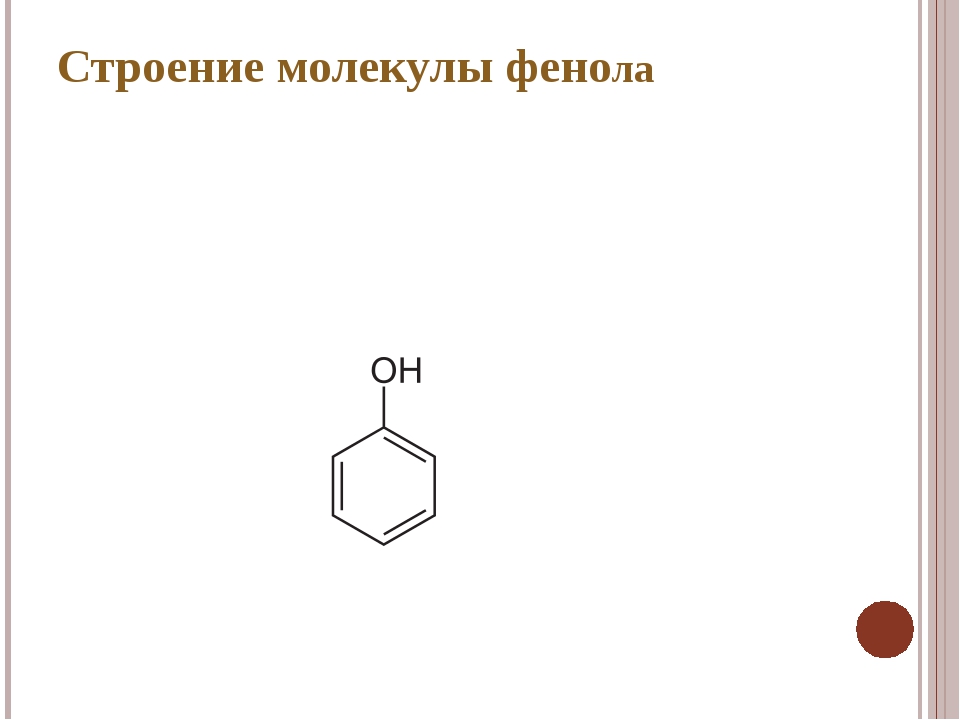
Анализ реактивности цистеина
in vitro в мышиной печени с 2-бутен-1,4-диал.( A ) Протеом печени мыши подвергали воздействию 2-бутен-1,4-диала (BDA) в начальных концентрациях в диапазоне от 1 мкМ до 3 мМ, а затем метили зондом IAyne (10 мкМ) с последующим клик-химическим придатком. родамин-азида для флуоресцентной визуализации (гель-ABPP) для измерения дозозависимого смещения зонда. ( B ) Анализ IsoTOP-ABPP протеома печени мыши после воздействия 10 мкМ 2-бутен-1,4-диала. Протеом печени мыши обрабатывали 2-бутен-1,4-диал и метили IAyne (100 мкМ) с последующим клик-химическим опосредованным придатком биотиновой метки, несущей последовательность расщепления протеазы TEV и изотопно легкий или тяжелый валин.Протеомы объединяли, обогащали авидином, расщепляли трипсином и выделяли модифицированные пептиды расщеплением TEV с последующим количественным протеомным анализом. Пептиды расположены в возрастающем соотношении легких (контрольных) и обработанных (тяжелых), а названия белков для пептидов, демонстрирующих соотношение легких и тяжелых >10, включены на рисунок. Пептиды, показывающие соотношение легких к тяжелым > 50, представлены как 50. ( C ) Анализ активности, показывающий, что инкубация чистого белка GAPDH с 2-бутен-1,4-диал (10 мкМ) ингибирует активность.* P < 0,05.
Пептиды, показывающие соотношение легких к тяжелым > 50, представлены как 50. ( C ) Анализ активности, показывающий, что инкубация чистого белка GAPDH с 2-бутен-1,4-диал (10 мкМ) ингибирует активность.* P < 0,05.
Среди более чем 600 идентифицированных модифицированных зондом пептидов мы идентифицировали 37 мишеней 2-бутен-1,4-диала (рис. 2 B и набор данных S1). Белки, связанные 2-бутен-1,4-диалом, имеют разнообразные функции и включают мишени, участвующие в биосинтезе белков, энергетическом метаболизме и биосинтезе стероидов. В частности, два из них содержали цистеины, которые соответствовали аннотированному каталитическому цистеину, C199 Nit1 (нитрилазоподобный белок 1) и C150 GAPDH (глицеральдегид-3-фосфатдегидрогеназа), что позволяет предположить, что функция этих ферментов была нарушена 2-бутен-1. ,4-циферб.Nit1 является белком, участвующим в регуляции апоптоза, поэтому ингибирование этого белка может привести к ускоренной пролиферации (31). GAPDH является гликолитическим ферментом, и его ингибирование, вероятно, влияет на гликолитический метаболизм и энергетику (32).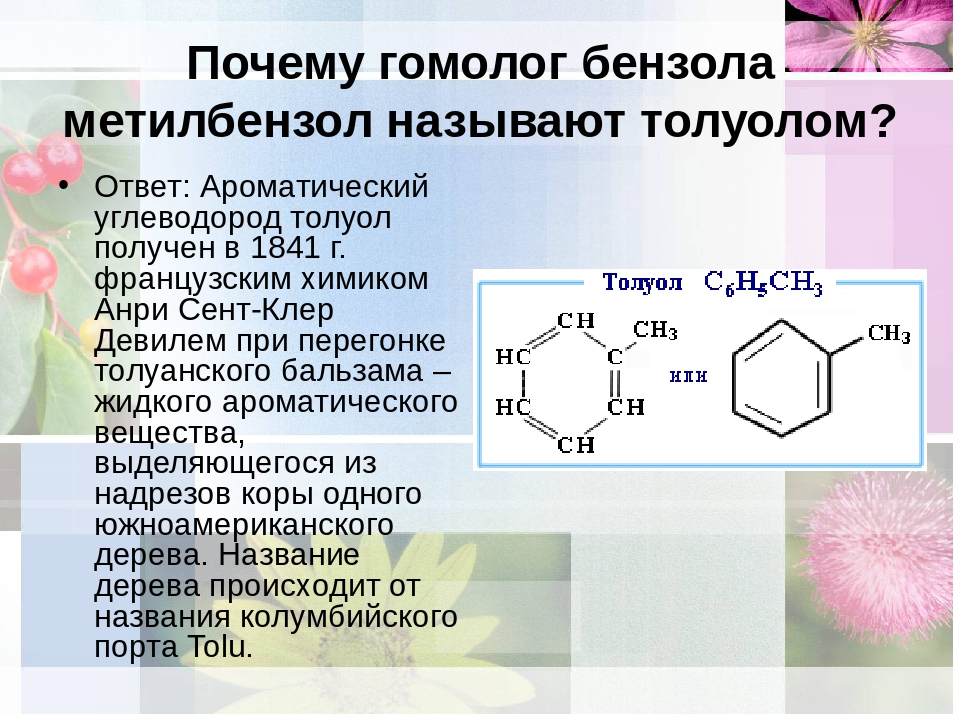 В соответствии с реактивностью с каталитическим C150 GAPDH мы показываем, что 2-бутен-1,4-диал ингибирует функцию GADPH в анализе активности субстрата (рис. 2 C ). Дальнейшая проверка целей, определенных с помощью IsoTOP-ABPP, и исследования токсикологии 2-бутен-1,4-диала in vivo могут дать представление о конечных точках воздействия на человека продуктов окисления фенола в питьевой воде.
В соответствии с реактивностью с каталитическим C150 GAPDH мы показываем, что 2-бутен-1,4-диал ингибирует функцию GADPH в анализе активности субстрата (рис. 2 C ). Дальнейшая проверка целей, определенных с помощью IsoTOP-ABPP, и исследования токсикологии 2-бутен-1,4-диала in vivo могут дать представление о конечных точках воздействия на человека продуктов окисления фенола в питьевой воде.
Для оценки потенциального образования других токсичных α,β-ненасыщенных альдегидов, образующихся при окислении обычных фенолсодержащих соединений, мы смоделировали обработку ряда метилированных фенолов процессом UV/H 2 O 2 . Мы полагались на образование уникальных продуктов реакции пирролин-2-она и пиррола энедиалей и оксеналей в присутствии NAL или GSH соответственно (рис. 3) (33). Для м-, o — и п-крезола воздействие •ОН, полученного в процессе УФ/Н 2 О 2 , приводило к образованию 2-бутен-1,4-диала и метилированного 2- аддукты бутен-1,4-диала (таблица 1 и SI Приложение , рис.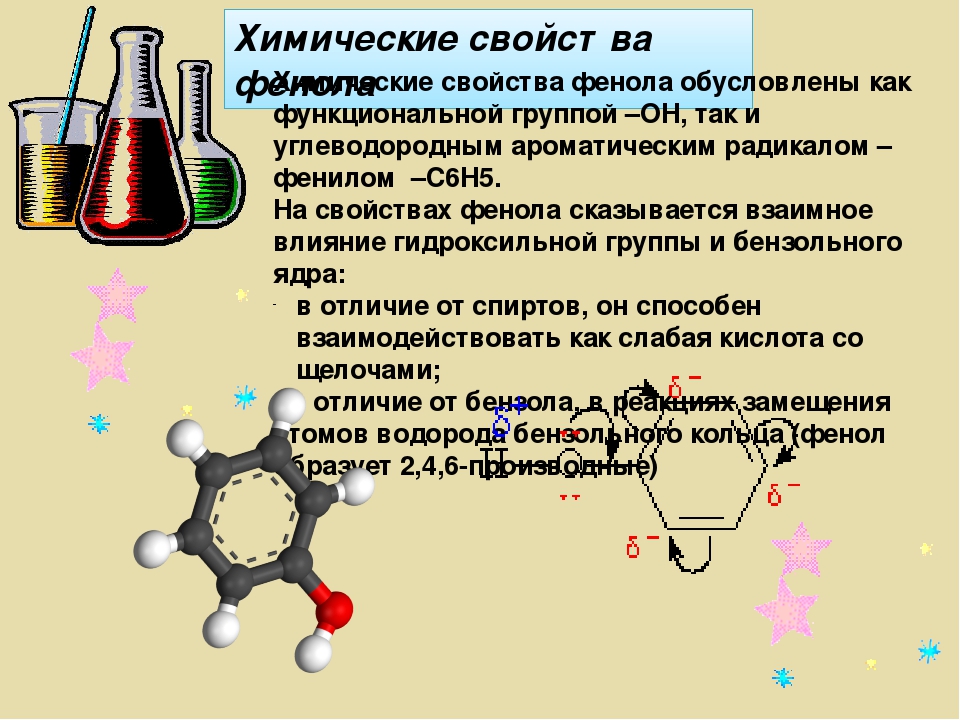 С5–С7). Последние были обнаружены в большем количестве как для п-, так и для м-крезола, тогда как для o -крезола преобладающим продуктом превращения альдегида был 2-бутен-1,4-диал (табл. 1). Точно так же окисление диметил- и триметилфенолов дает аддукты NAL и GSH, соответствующие присутствию одной, двух или трех дополнительных метильных групп по сравнению с продуктом трансформации, наблюдаемым при окислении фенола ( SI, Приложение , рис. S10). Таким образом, наши данные показывают, что окисление алкилзамещенных фенолов по процессу UV/H 2 O 2 обычно приводит к образованию α,β-ненасыщенных эндиалов и оксеналей.
С5–С7). Последние были обнаружены в большем количестве как для п-, так и для м-крезола, тогда как для o -крезола преобладающим продуктом превращения альдегида был 2-бутен-1,4-диал (табл. 1). Точно так же окисление диметил- и триметилфенолов дает аддукты NAL и GSH, соответствующие присутствию одной, двух или трех дополнительных метильных групп по сравнению с продуктом трансформации, наблюдаемым при окислении фенола ( SI, Приложение , рис. S10). Таким образом, наши данные показывают, что окисление алкилзамещенных фенолов по процессу UV/H 2 O 2 обычно приводит к образованию α,β-ненасыщенных эндиалов и оксеналей.
Общий механизм реакции окисления метилированных фенолов УФ/Н 2 O 2 с образованием оксеналей и энедиалов. R 1 , R 2 и R 3 указывают расположение остатков -H или -CH 3 в C4-дикарбонильных соединениях, образующихся при окислении, и зависят от расположения метильных заместителей в исследуемых соединениях. фенолы (табл. 1).
фенолы (табл. 1).
Продукты и относительные выходы окисления метилзамещенных фенолов
Повсеместное присутствие •ОН в живых организмах (34), природных водах (35⇓–37), тумане (38) и атмосфере ( 39) означает, что наши наблюдения могут иметь потенциальное значение, выходящее за рамки окислительной обработки воды.Окислительный стресс, вызванный активными формами кислорода, играет важную роль в естественных условиях и связан с различными неблагоприятными последствиями для здоровья, включая болезнь Паркинсона и сердечно-сосудистые заболевания (34). Например, реакция •ОН с фенольной аминокислотой тирозином приводит к образованию реакционноспособных промежуточных соединений, способных реагировать с другими биомолекулами, что приводит к перекрестным связям белок-белок и белок-ДНК (40). Кроме того, реакции аминокислот, таких как тирозин, с •OH в освещенных солнцем поверхностных водах связаны с образованием гуминоподобных веществ, которые играют важную роль в экосистемах поверхностных вод, в частности, для геохимического круговорота микроэлементов ( 41). В тропосфере эти реакции также участвуют в формировании вторичных органических аэрозолей (42). Образование реактивных продуктов превращения тирозина при воздействии •ОН до сих пор связывали исключительно с образованием тирозильных радикалов с углеродным центром (43). Однако наши результаты раскрывают ранее неизвестный механизм реакции. При воздействии на раствор N -ацетилтирозина в • ОН мы продемонстрировали образование продуктов кросс-сочетания по механизму, аналогичному наблюдаемому для замещенных фенолов, через образование продукта эндиального превращения (рис.4).
В тропосфере эти реакции также участвуют в формировании вторичных органических аэрозолей (42). Образование реактивных продуктов превращения тирозина при воздействии •ОН до сих пор связывали исключительно с образованием тирозильных радикалов с углеродным центром (43). Однако наши результаты раскрывают ранее неизвестный механизм реакции. При воздействии на раствор N -ацетилтирозина в • ОН мы продемонстрировали образование продуктов кросс-сочетания по механизму, аналогичному наблюдаемому для замещенных фенолов, через образование продукта эндиального превращения (рис.4).
Окисление N -ацетил-тирозина в системе УФ/Н 2 O 2 . ( A ) Окисление N -ацетил-тирозина с помощью • OH и УФ-света и последующая реакция образовавшегося 1,4-ендиала с N -(α)-ацетил-лизином (NAL) и N -ацетилцистеин (NAC) и NAL (спектры аддуктов MS 2 приведены в SI Приложение , рис. S12). ( B ) Предлагаемый путь реакции реакционноспособного продукта трансформации энедиала N -ацетилтирозина и его реакции с NAL и NAC+NAL.
Ранее опубликованные исследования показывают, что дигидроксибензолы и органические кислоты составляют значительную долю продуктов трансформации, образующихся при воздействии на фенолы • ОН или УФ-света (5, 6). Наши результаты показывают, что в этом процессе также образуется неизвестная ранее группа токсичных соединений — энедиалы и оксенали, хотя и с меньшим выходом, чем другие продукты. Токсичность α,β-ненасыщенных карбонильных соединений, в частности α,β-ненасыщенных альдегидов, объясняется их высокой реакционной способностью за счет нуклеофильного присоединения как к карбонильному, так и к β-углероду (44).В результате α,β-ненасыщенные альдегиды гораздо более токсичны, чем их насыщенные аналоги (18, 45).
Помимо фенола, в окружающей среде присутствуют различные природные и антропогенные фенольные соединения. В дополнение к присутствию фенольных групп во многих промышленных химикатах метилфенолы представляют собой важные фрагменты водного природного органического вещества (46).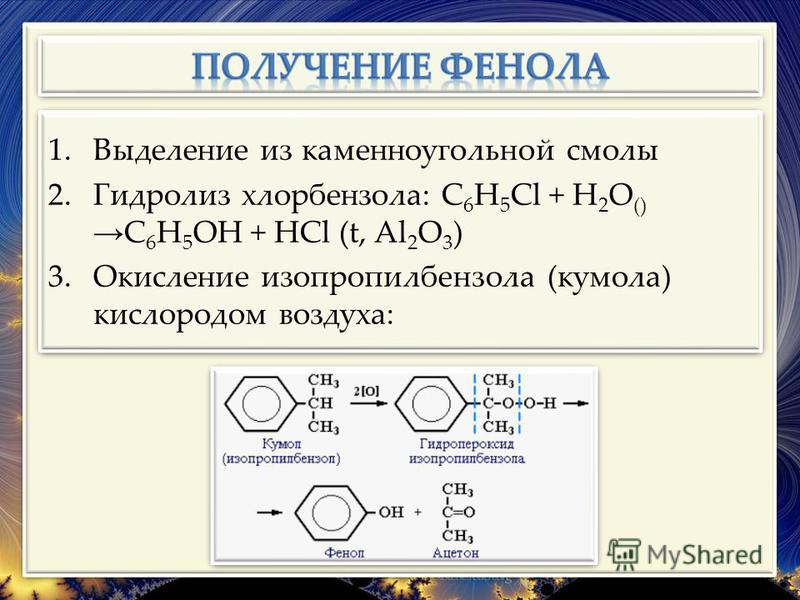 Кроме того, фенольные соединения выделяются в результате процессов горения и попадают в поверхностные воды в основном в результате влажного и сухого осаждения (47⇓–49).Таким образом, использование технологий обработки на основе •OH и УФ-обработки может привести к образованию многих различных типов α,β-ненасыщенных альдегидов, которые могут реагировать с биомолекулами (44).
Кроме того, фенольные соединения выделяются в результате процессов горения и попадают в поверхностные воды в основном в результате влажного и сухого осаждения (47⇓–49).Таким образом, использование технологий обработки на основе •OH и УФ-обработки может привести к образованию многих различных типов α,β-ненасыщенных альдегидов, которые могут реагировать с биомолекулами (44).
Судьба диальдегидов в технических системах очистки и распределения питьевой воды зависит от процессов очистки воды, которые следуют за процессом окислительной обработки. Потери при улетучивании из открытых бассейнов, вероятно, будут небольшими, поскольку диальдегиды не особенно летучи (например, концентрация стандартного 2-бутен-1,4-диала уменьшилась менее чем на 10% через 2 дня в открытом контейнере, хранящемся при комнатной температуре). температура).Из-за полярной природы диальдегидов удаление путем сорбции активированным углем также, вероятно, будет небольшим в условиях, используемых на станциях очистки питьевой воды. Соединения также не будут подвергаться фотолизу в заметной степени после их образования (например, концентрации 2-бутен-1,4-диала не уменьшались после удаления большей части фенола в экспериментах, изображенных на рис. 1). Таким образом, трансформация на последующих стадиях, когда вода подвергается воздействию микробов (например, при фильтрации песком или биологическом активированном угле) или в системе распределения питьевой воды, вероятно, является наиболее важным стоком для энедиальных и оксенальных видов.Необходимы дополнительные исследования для оценки возникновения и скорости этих процессов удаления.
Соединения также не будут подвергаться фотолизу в заметной степени после их образования (например, концентрации 2-бутен-1,4-диала не уменьшались после удаления большей части фенола в экспериментах, изображенных на рис. 1). Таким образом, трансформация на последующих стадиях, когда вода подвергается воздействию микробов (например, при фильтрации песком или биологическом активированном угле) или в системе распределения питьевой воды, вероятно, является наиболее важным стоком для энедиальных и оксенальных видов.Необходимы дополнительные исследования для оценки возникновения и скорости этих процессов удаления.
Таким образом, мы показываем, что образование аддуктов между аминокислотами и белками может быть использовано в качестве чувствительного средства идентификации продуктов реактивного превращения, образующихся при окислительной обработке воды. Для реакции фенолов с •ОН и УФ-светом это привело к неожиданному открытию продуктов превращения оксеналей и эндиалов. Чувствительные аналитические подходы, подобные описанным здесь, предлагают мощный подход для идентификации реактивных электрофилов.
